The Pulm PEEPs are joined again by Natalie West to discuss a patient who presented with a chronic, productive cough. Listen in today as we work through our differential diagnosis, interpret basic pulmonary testing, and share our clinical reasoning along the way. We have some fantastic diagnostic and treatment teaching points, so once you’ve solved the case check out the takeaways and infographics below. Please let us know any additional insights you have on Twitter!

Patient Presentation
A 50-year-old woman, who is a never smoker, with a past medical history of recurrent pancreatitis presents to the pulmonary clinic with a chronic, productive cough. Her cough has been present for 3 years and has increased in frequency to now being present daily. In the last three months, the cough has also worsened and is productive of small amounts of yellow to green sputum. She has a history of chronic post-nasal drip and sinus infections, and uses intranasal steroids, but has not noted changes in these symptoms. There is no significant family history of pulmonary disease, and an exposure history review of symptoms is negative.
On physical exam, she was a thin woman who appeared her stated age and was breathing comfortably on room air. Her exam was notable for mild expiratory wheezing, primarily on auscultation of the right posterior lung field. She had no cyanosis, clubbing, evidence of volume overload, or abdominal tenderness.
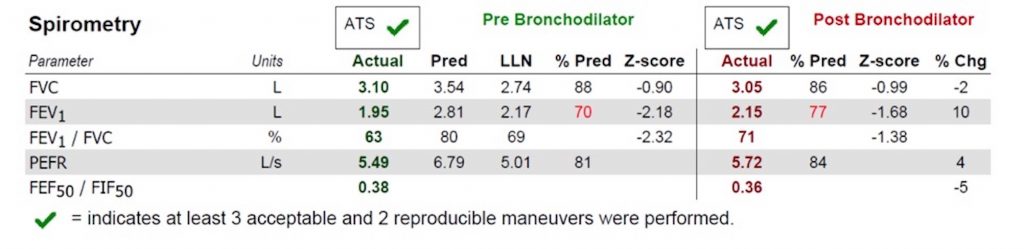
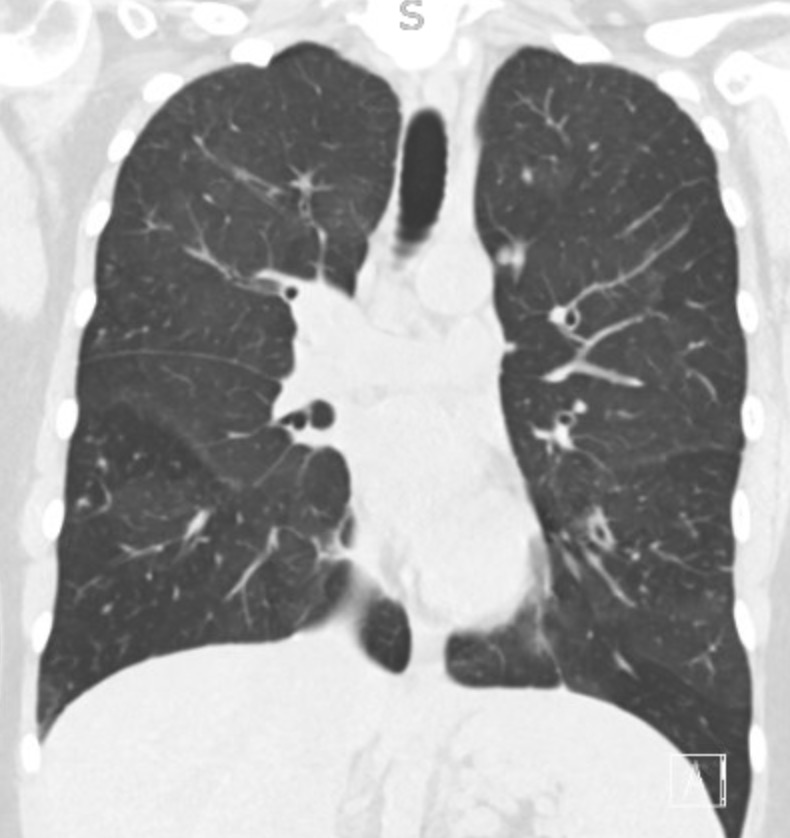
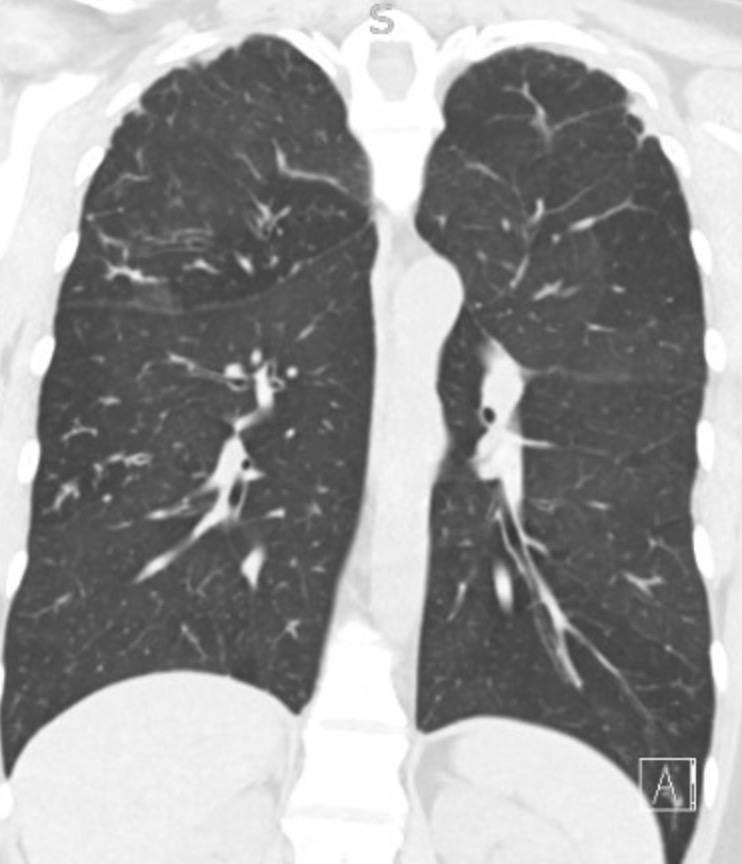
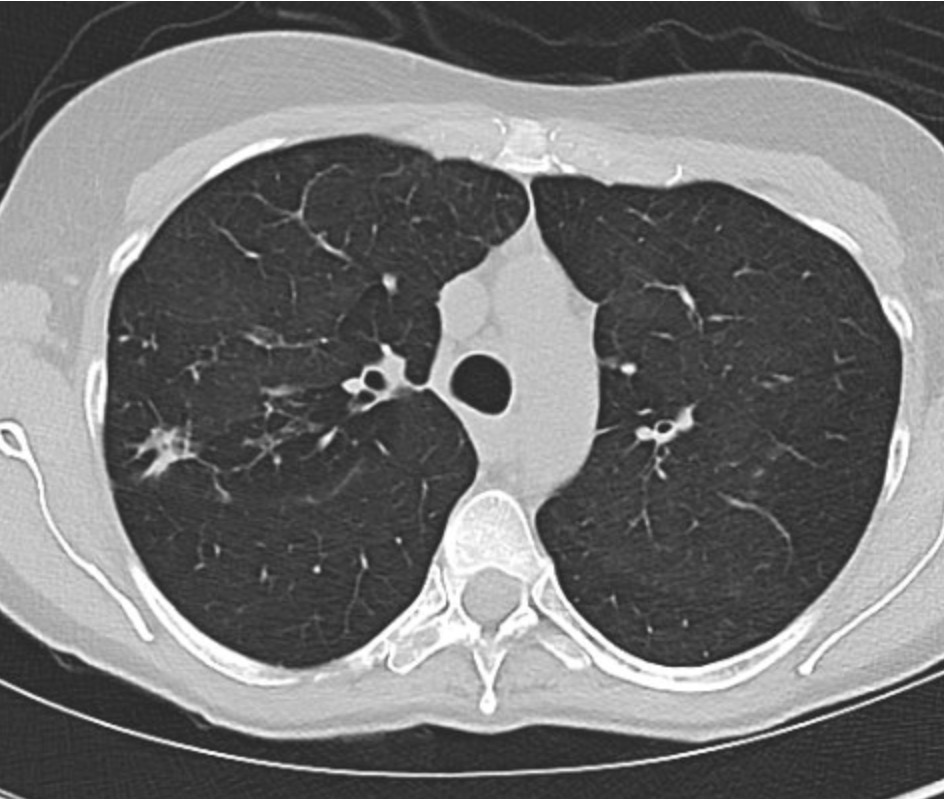
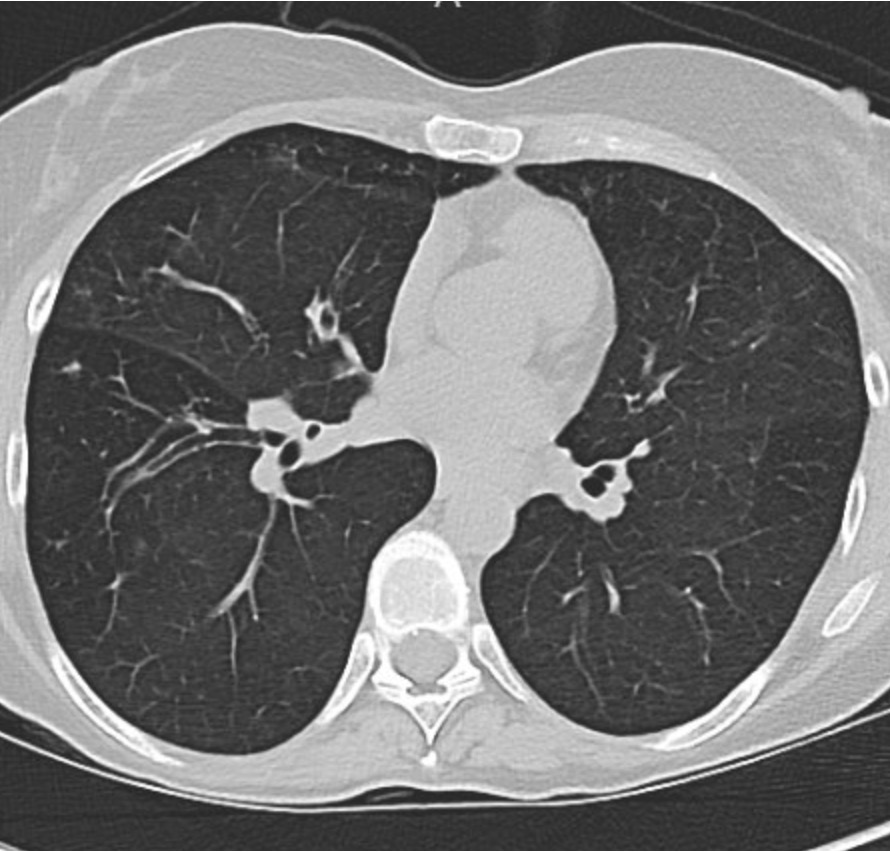
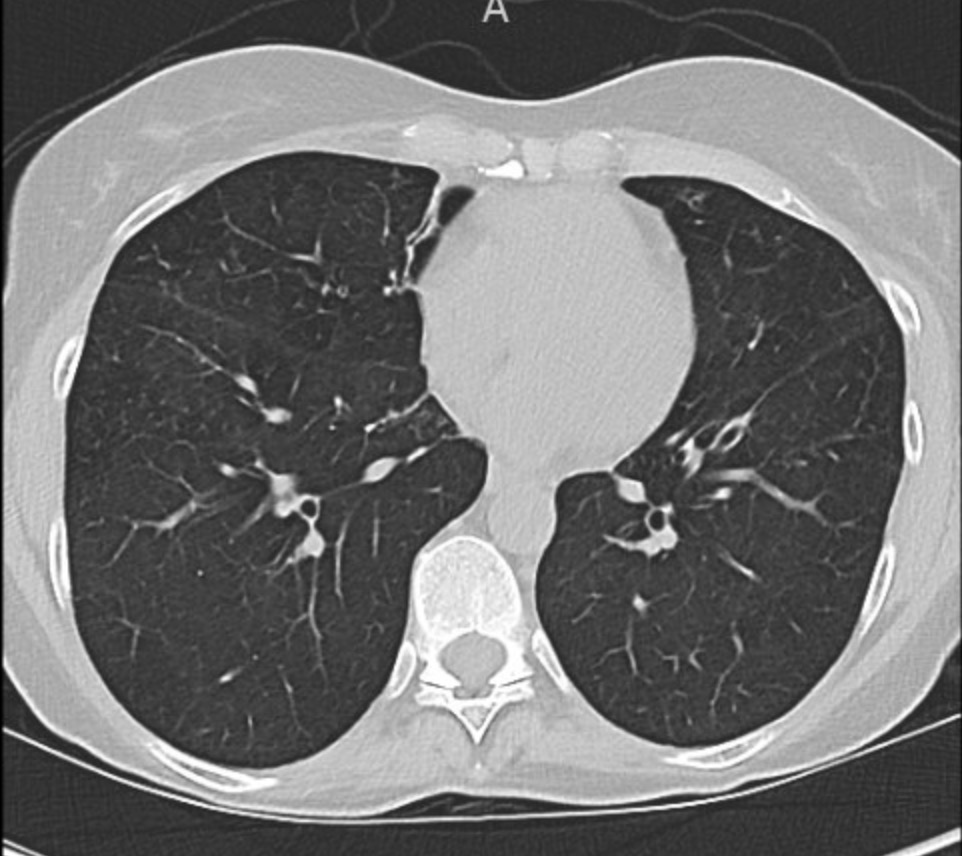
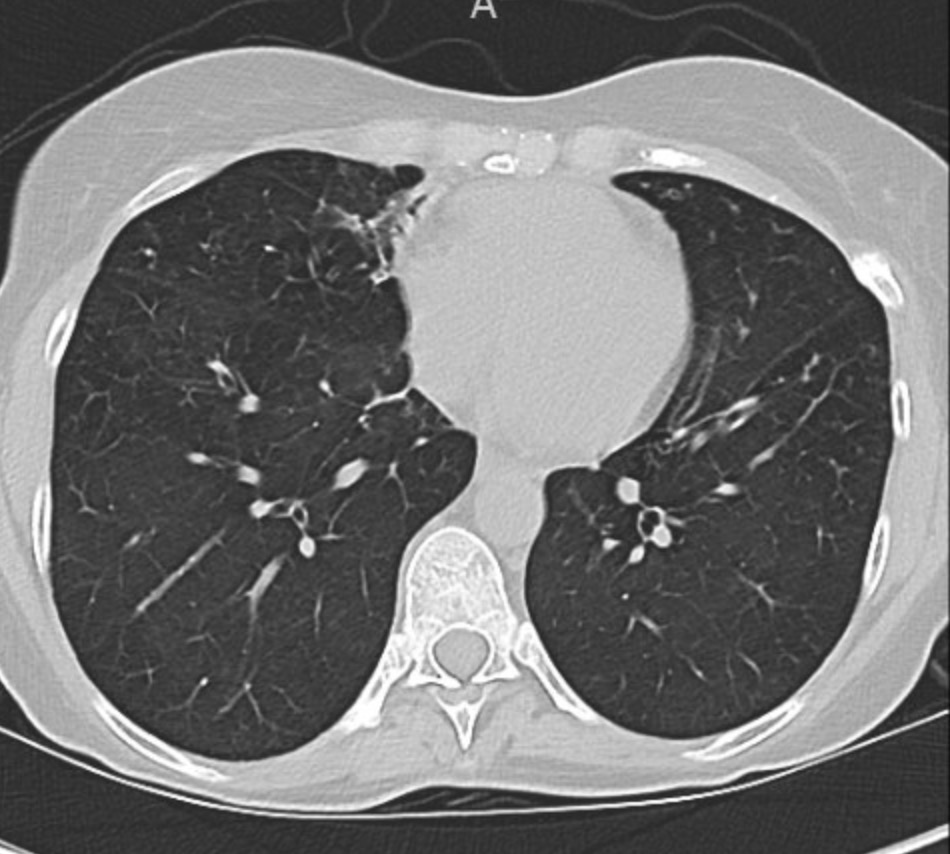
Key Learning Points
**Spoilers Ahead** If you want to think through the case on your own we advise listening to the episode first before looking at the infographics below.
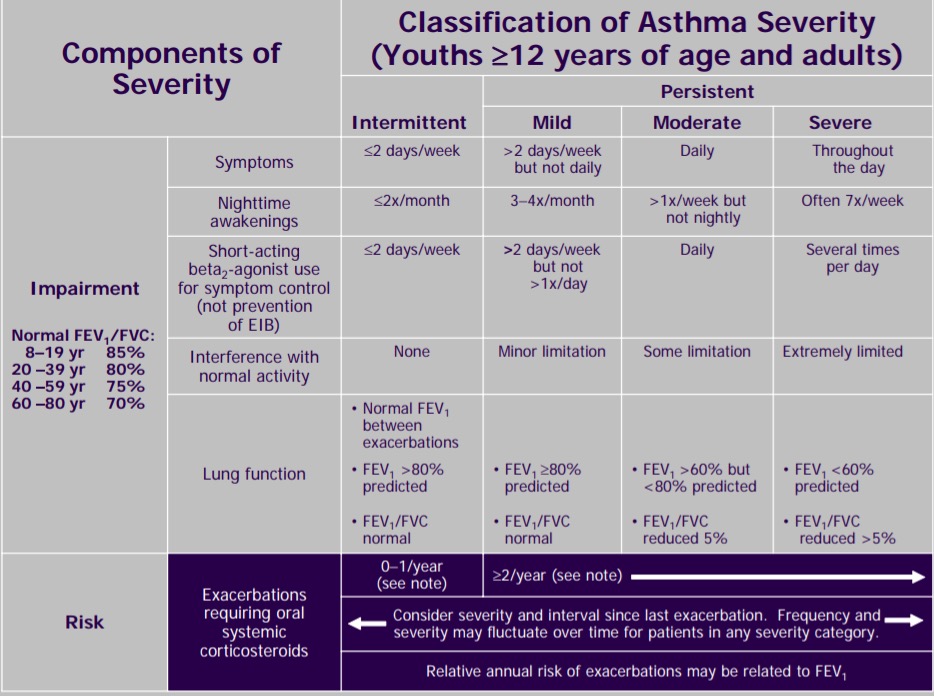
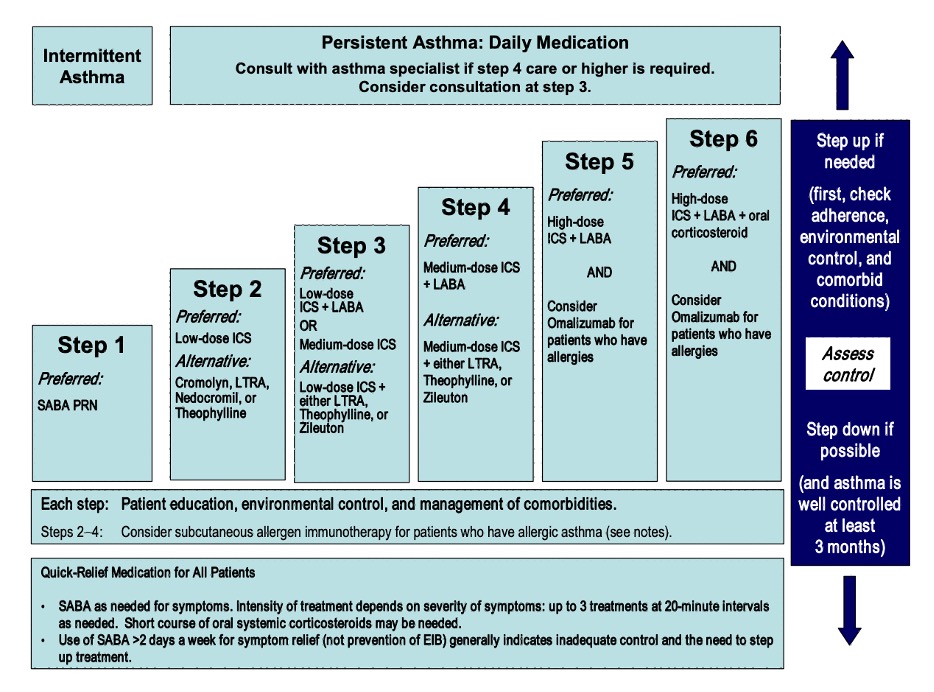
Differential Diagnosis of Chronic Cough
Three most common causes: upper airway cough syndrome, GERD, cough variant asthma
Additional etiologies to consider: chronic bronchitis, post-infectious after a respiratory tract infection, bronchiectasis, ACE inhibitors, lung cancer, eosinophilic bronchitis, interstitial lung disease
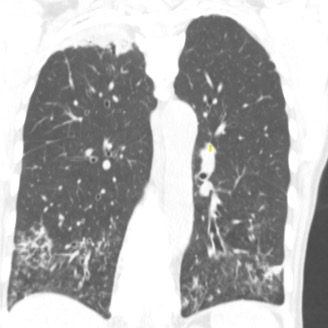
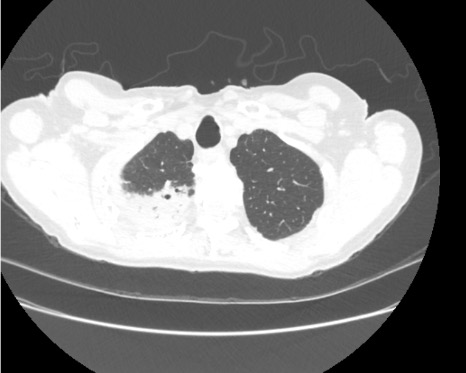
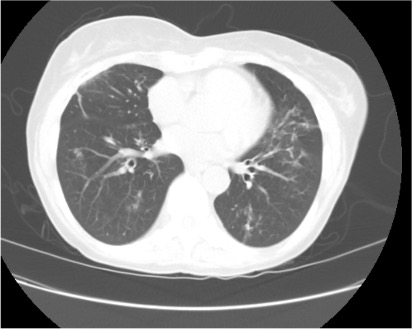
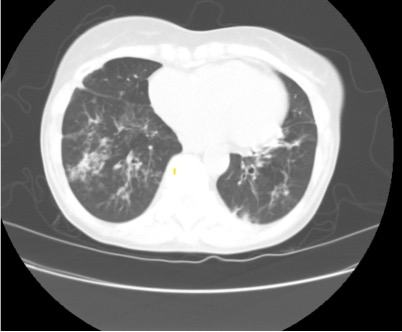
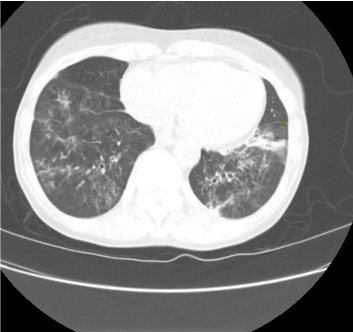
Imaging Pearl
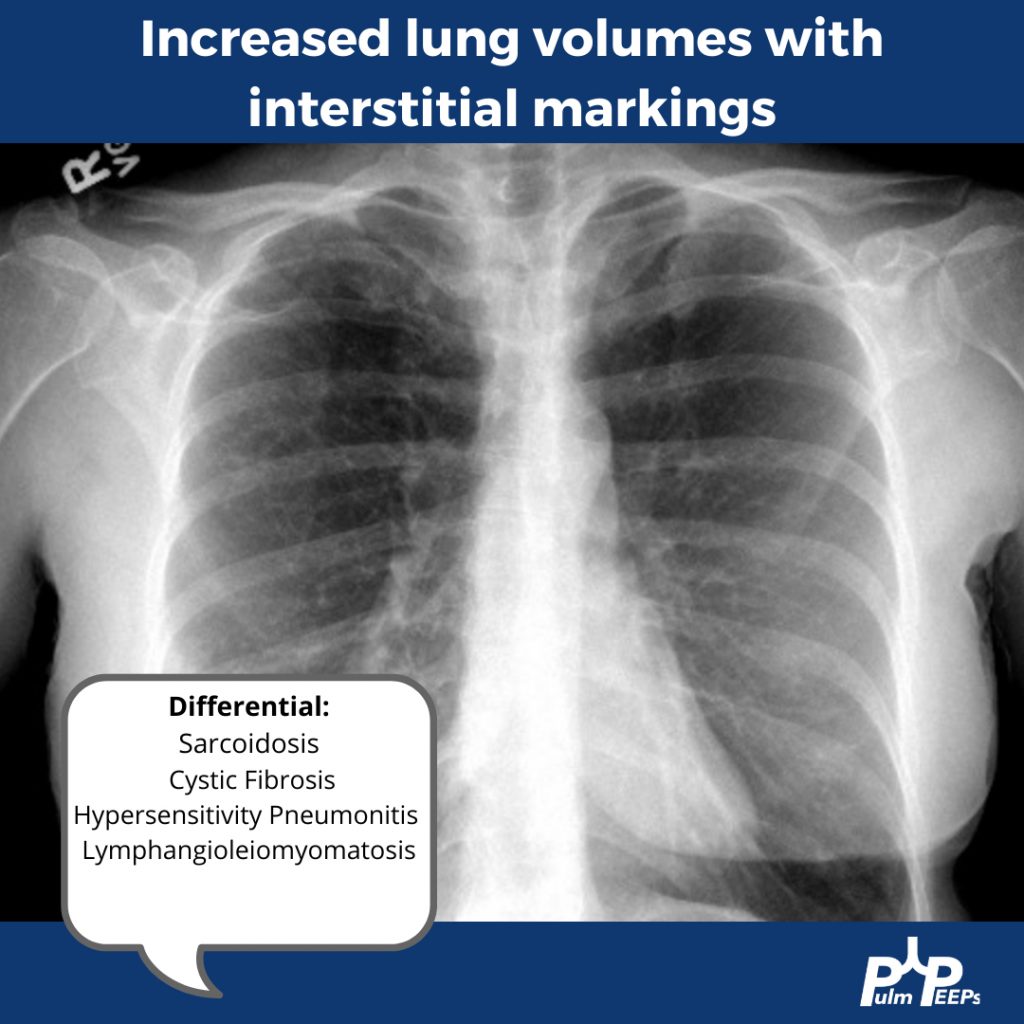
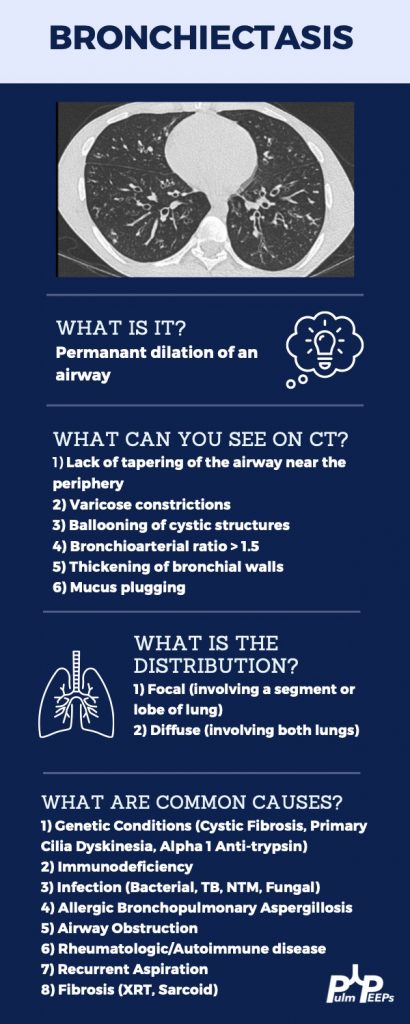
Evaluating Bronchiectasis

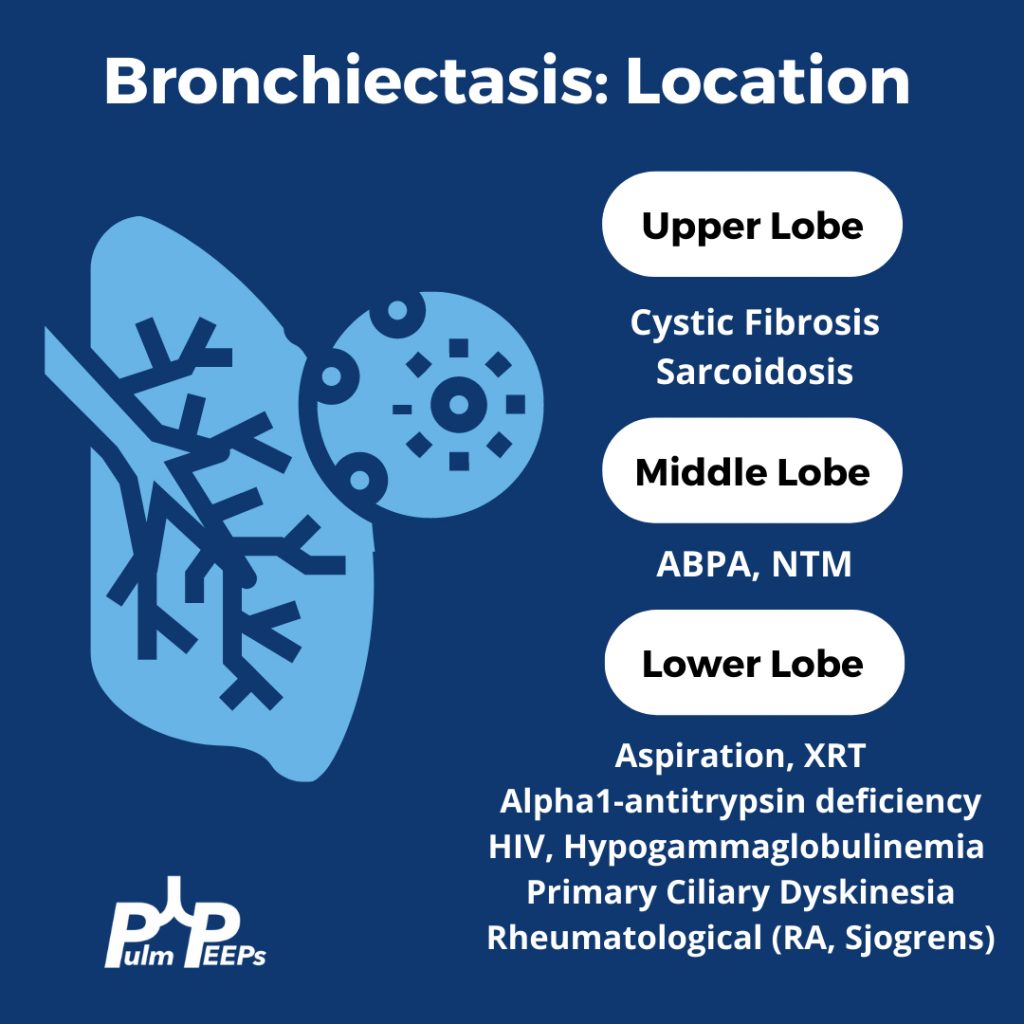
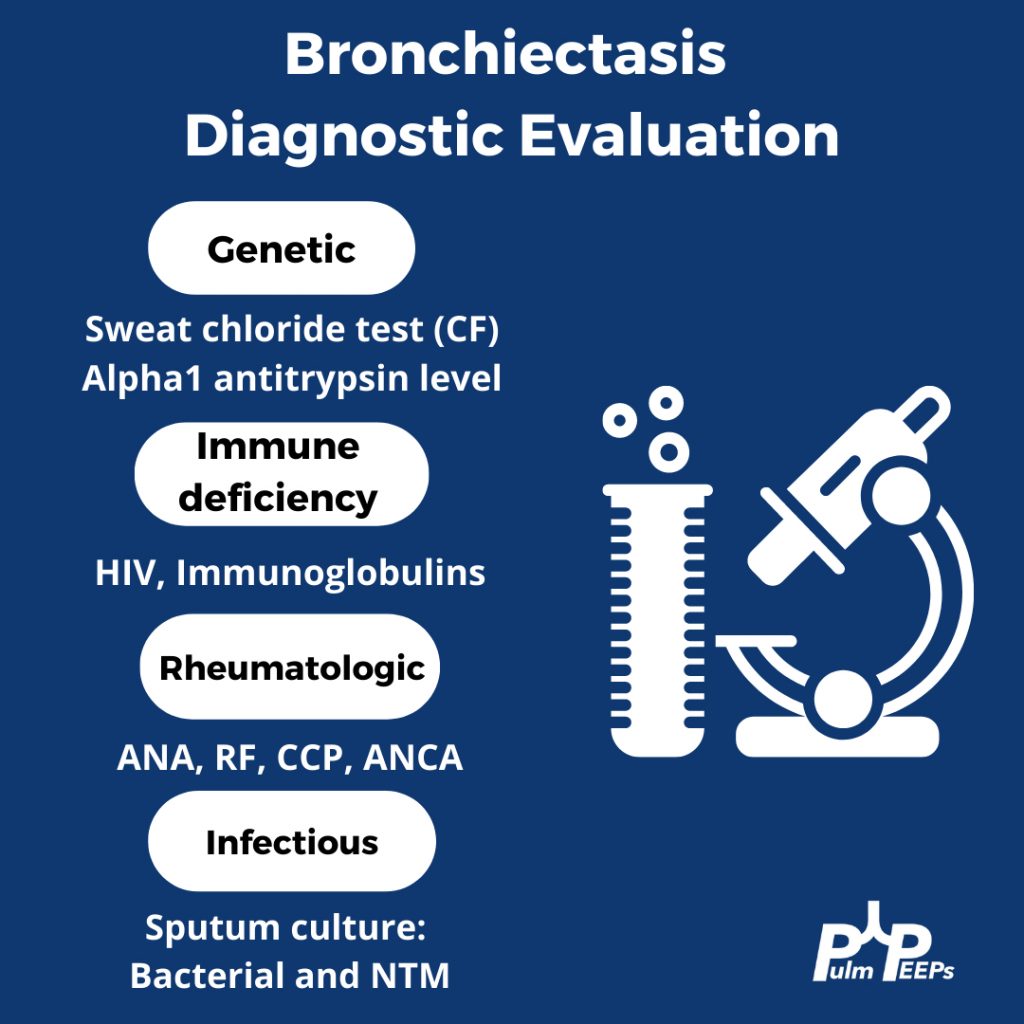
Making a New Diagnosis of Cystic Fibrosis in an Adult
Sweat testing
Sweat testing should be done in CF accredited center. Inform patients that there are no needles involved. Pilocarpine and electrical stimulation are applied to the arm or leg to stimulate the sweat gland, and then sweat is collected on filter paper, a gauze, or a plastic coil. From there, the amount of chloride in the sweat is calculated
Results
< 30 normal
31 – 60 indeterminate
> 60 is positive and Cystic Fibrosis is likely
What do you do with an Indeterminate test?
Patients with milder phenotypes of Cystic Fibrosis can have a normal or indeterminate sweat chloride level, and 10% of adults diagnosed with CF have a normal sweat chloride. If the sweat chloride test is indeterminate or normal, but suspicion is high for CF, then genetic testing for the whole array of mutations should be performed

References and links for further reading
- Morice AH, Millqvist E, Bieksiene K, et al. ERS guidelines on the diagnosis and treatment of chronic cough in adults and children. European Respiratory Journal. 2020;55(1). doi:10.1183/13993003.01136-2019
- Barker AF. Bronchiectasis. New England Journal of Medicine. 2002;346(18):1383-1393. doi:10.1056/NEJMra012519
- Bronchiectasis: a case-based approach to investigation and management | European Respiratory Society. Accessed November 23, 2021. https://err.ersjournals.com/content/27/149/180016
- Rowe SM, Miller S, Sorscher EJ. Cystic Fibrosis. New England Journal of Medicine. 2005;352(19):1992-2001. doi:10.1056/NEJMra043184
- Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. The Lancet. 2021;397(10290):2195-2211. doi:10.1016/S0140-6736(20)32542-3
- Jain R. Diagnosing Cystic Fibrosis in Adults: Better Late Than Never. Annals ATS. 2018;15(10):1140-1141. doi:10.1513/AnnalsATS.201806-432ED
Podcast: Play in new window | Download
Subscribe: Apple Podcasts | Spotify | Amazon Music | Android | iHeartRadio | Podcast Index | RSS