We are excited to start the month off with a new #RadiologyRounds
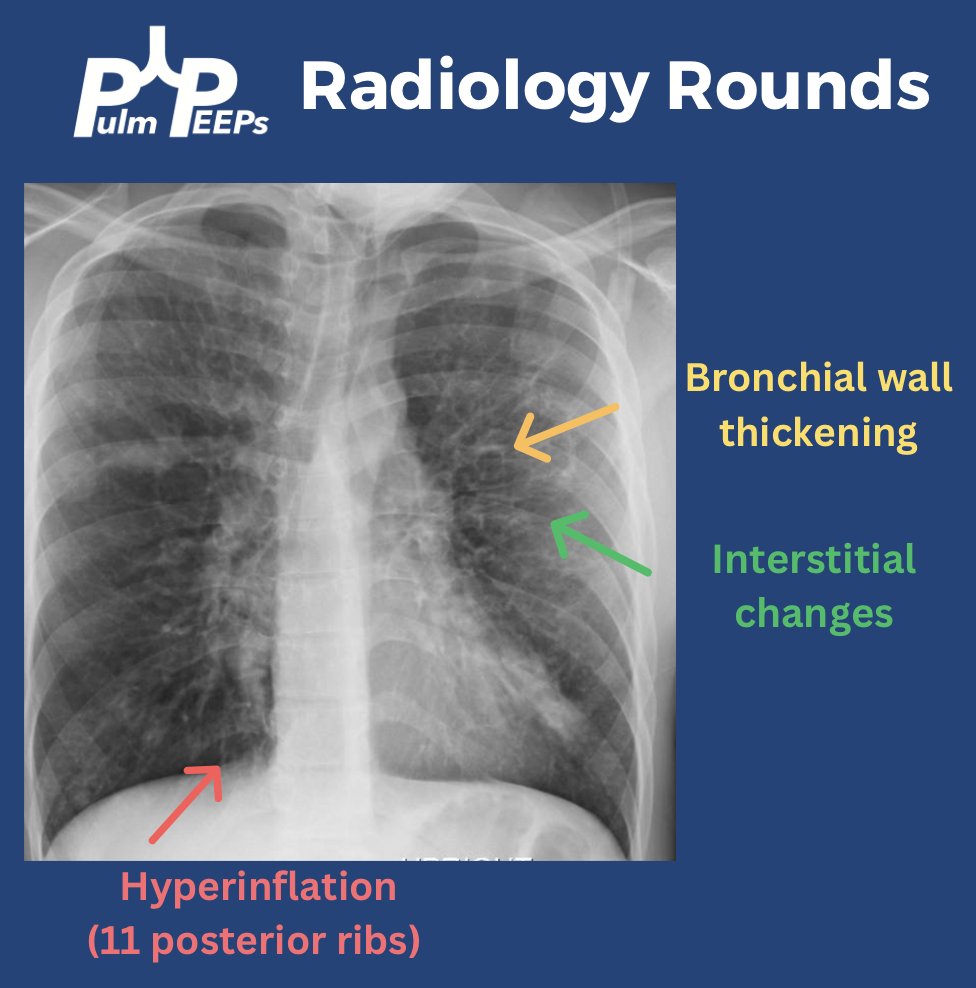
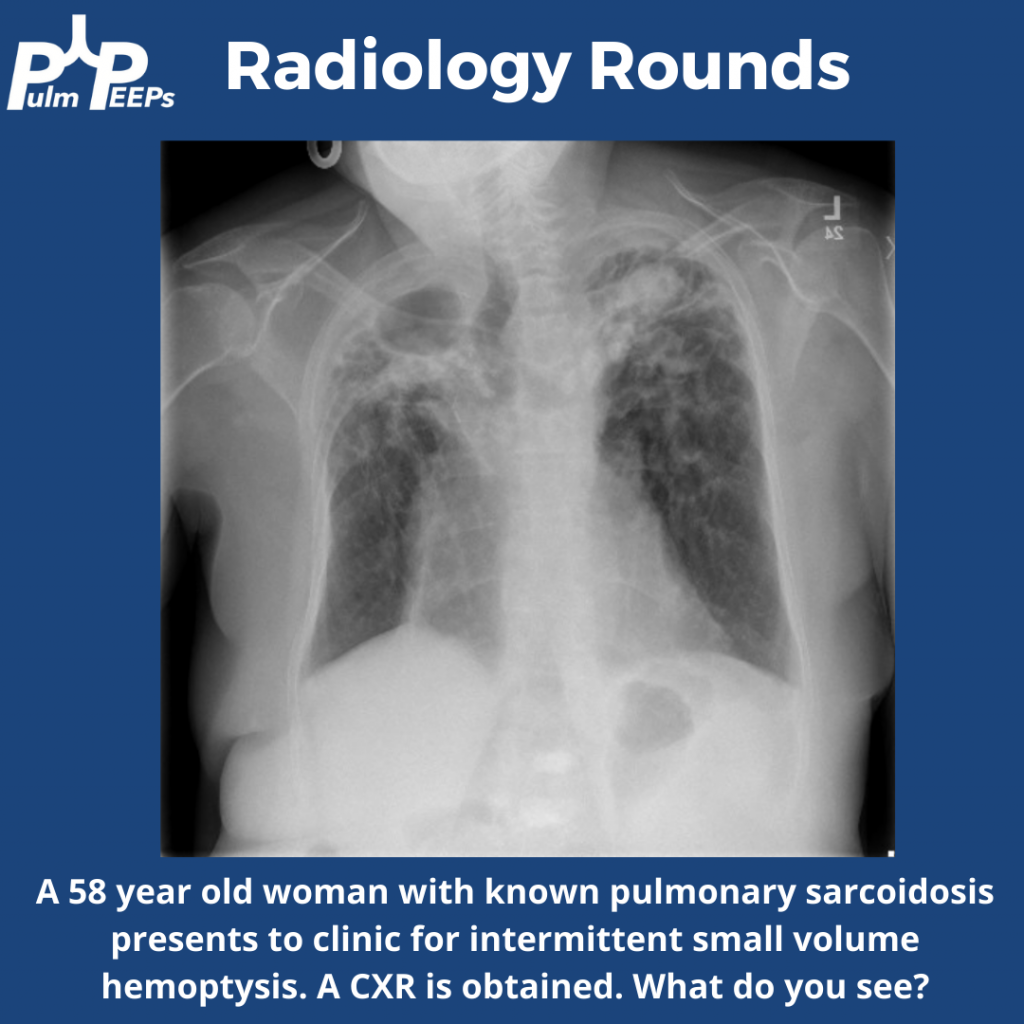
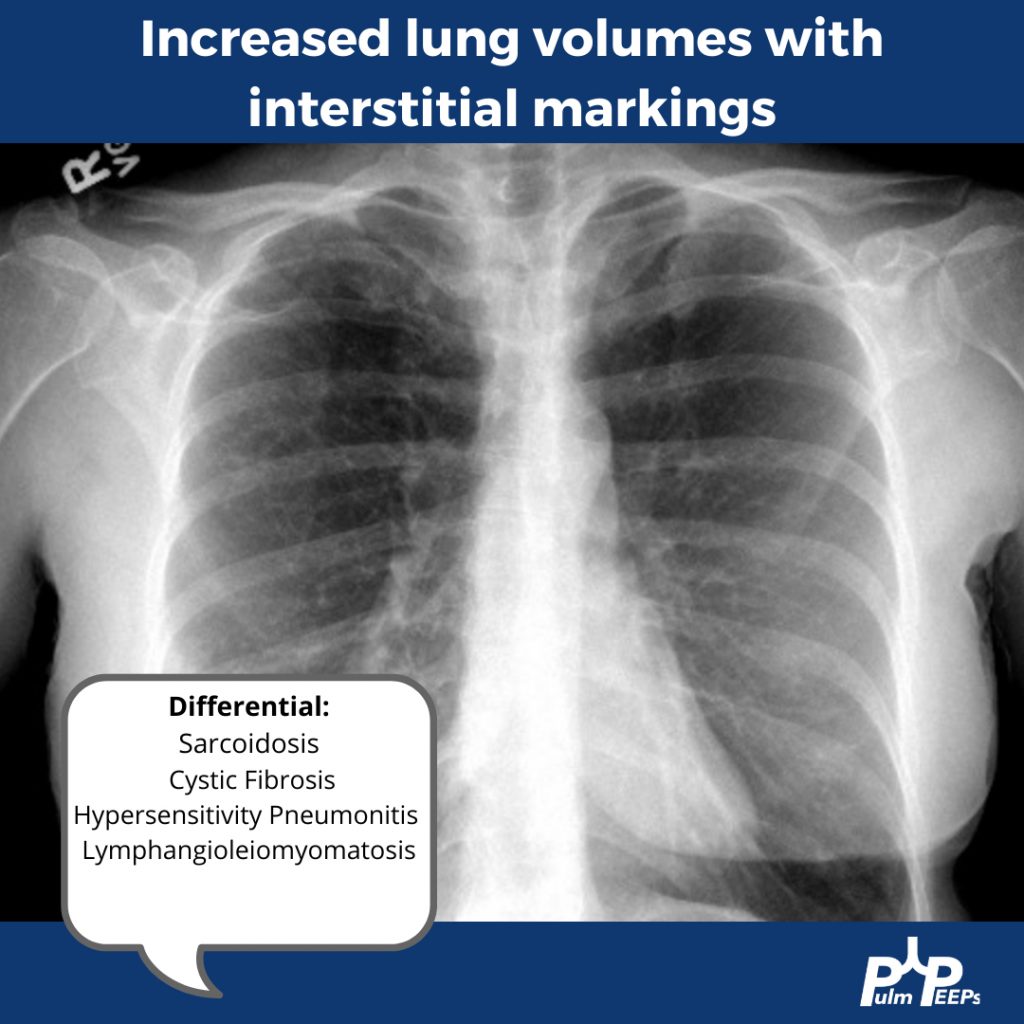
A young adult man in his 20s presents with dyspnea on exertion, productive cough, intermittent wheezing and general fatigue. A chest x-ray was obtained as part of his work-up.
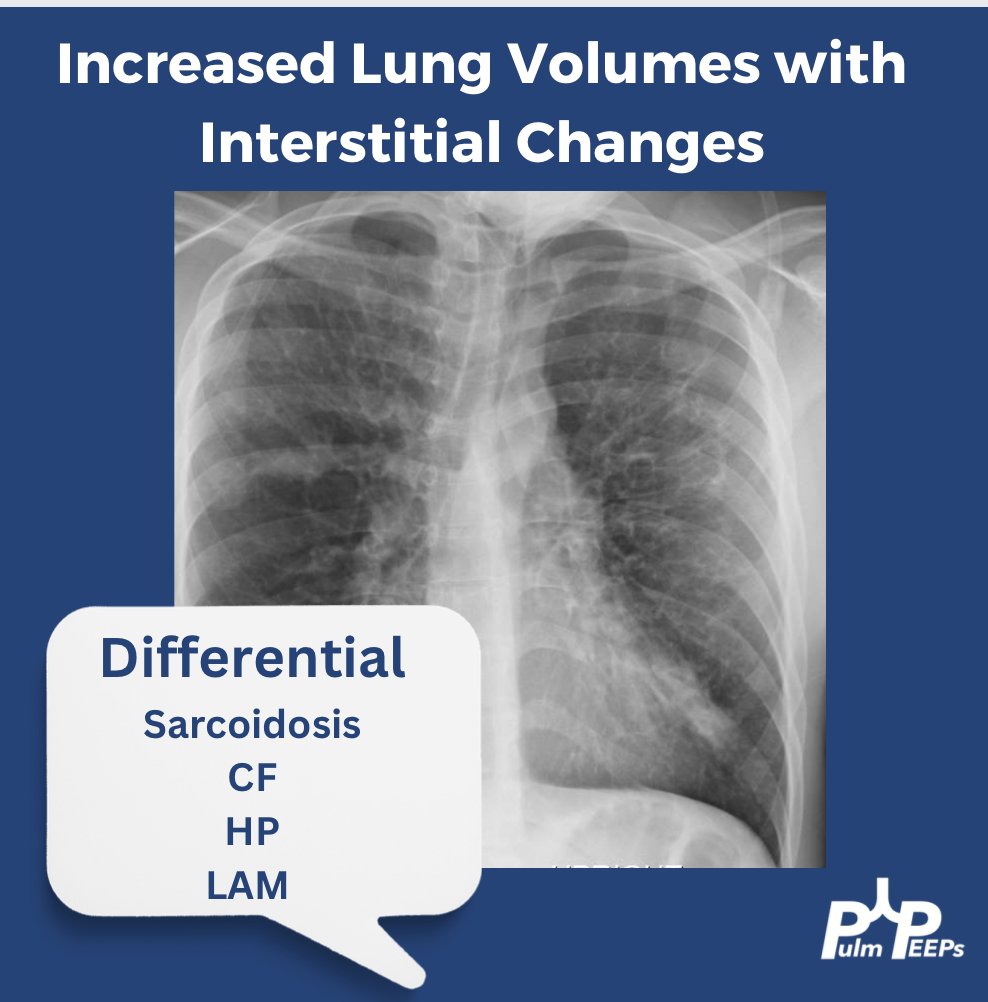
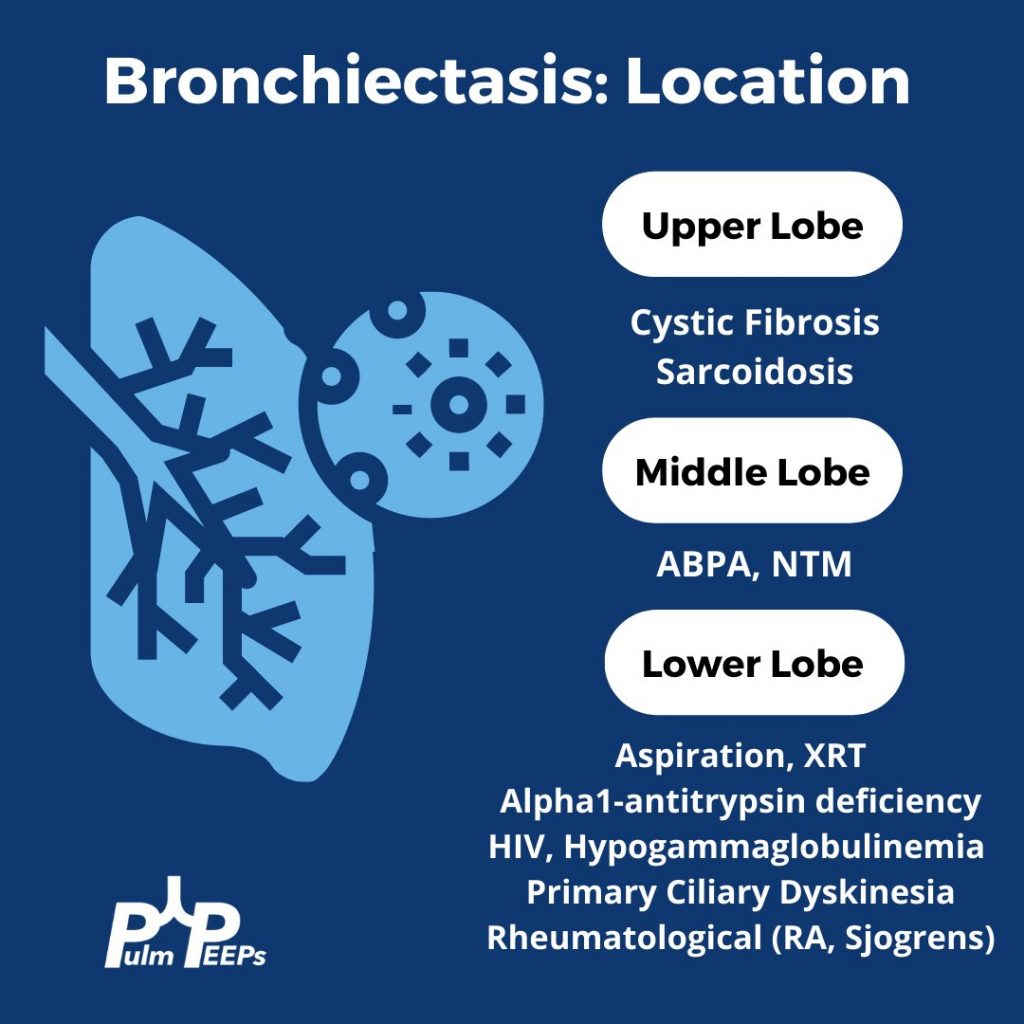
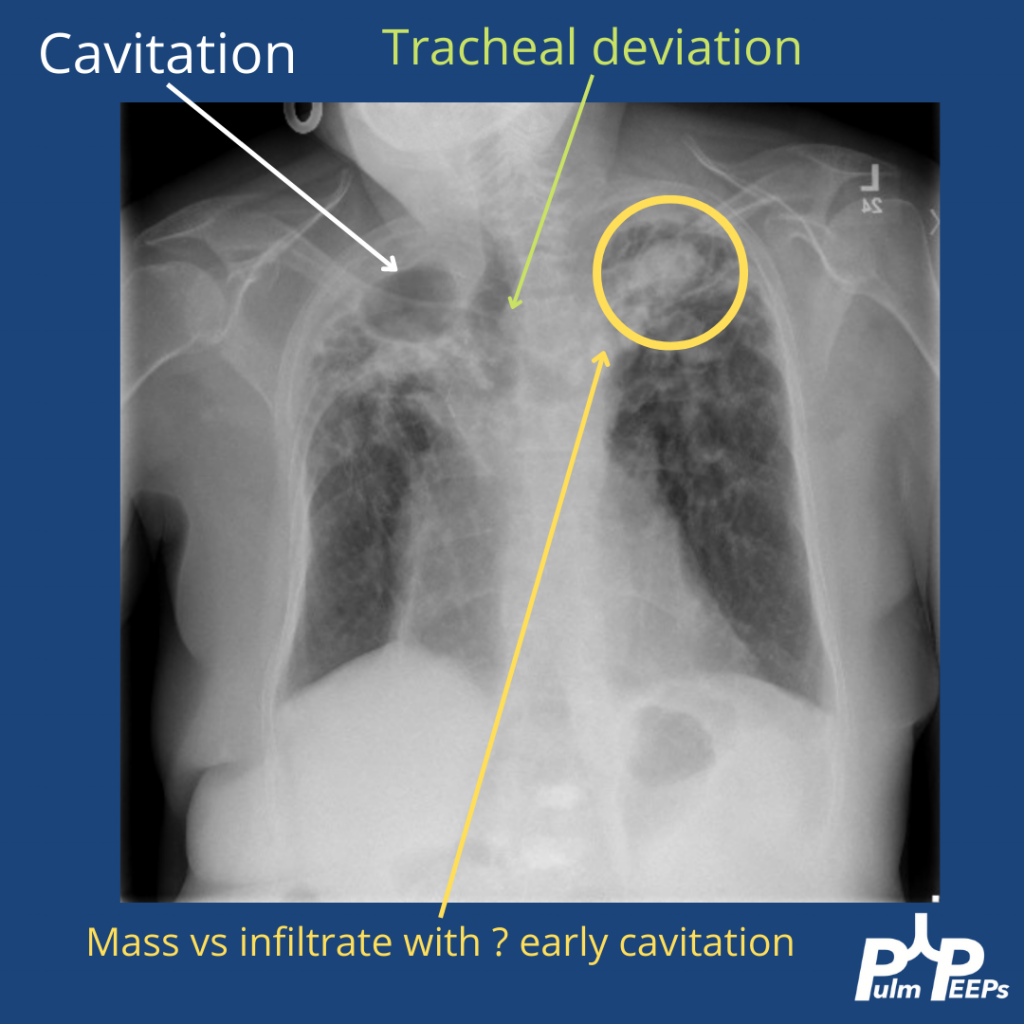
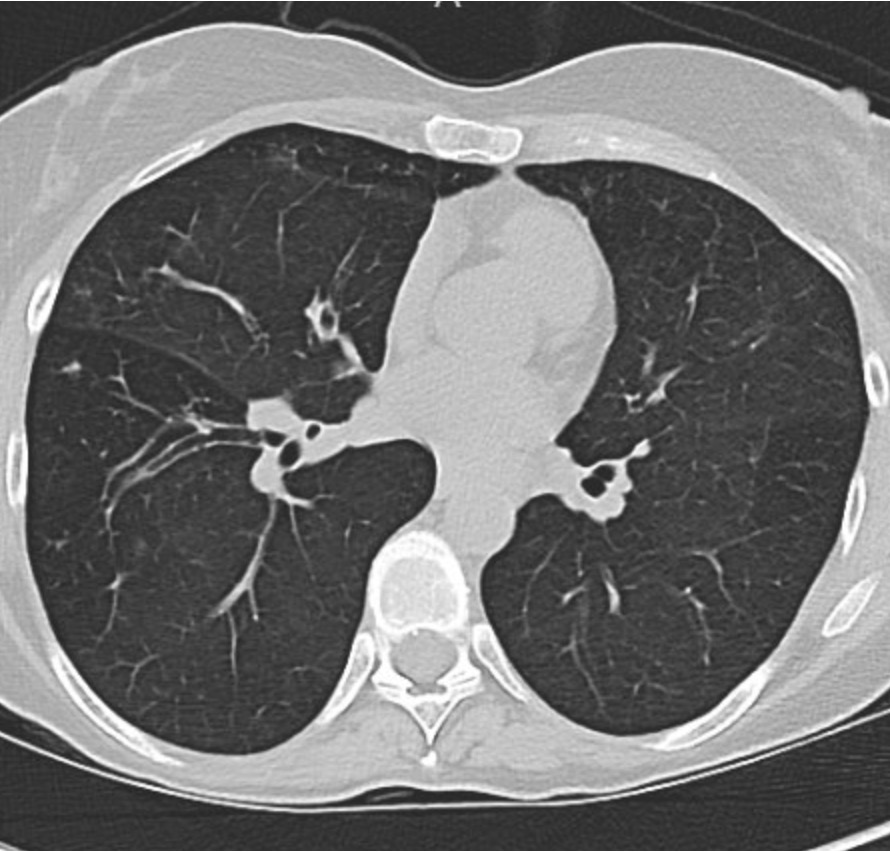
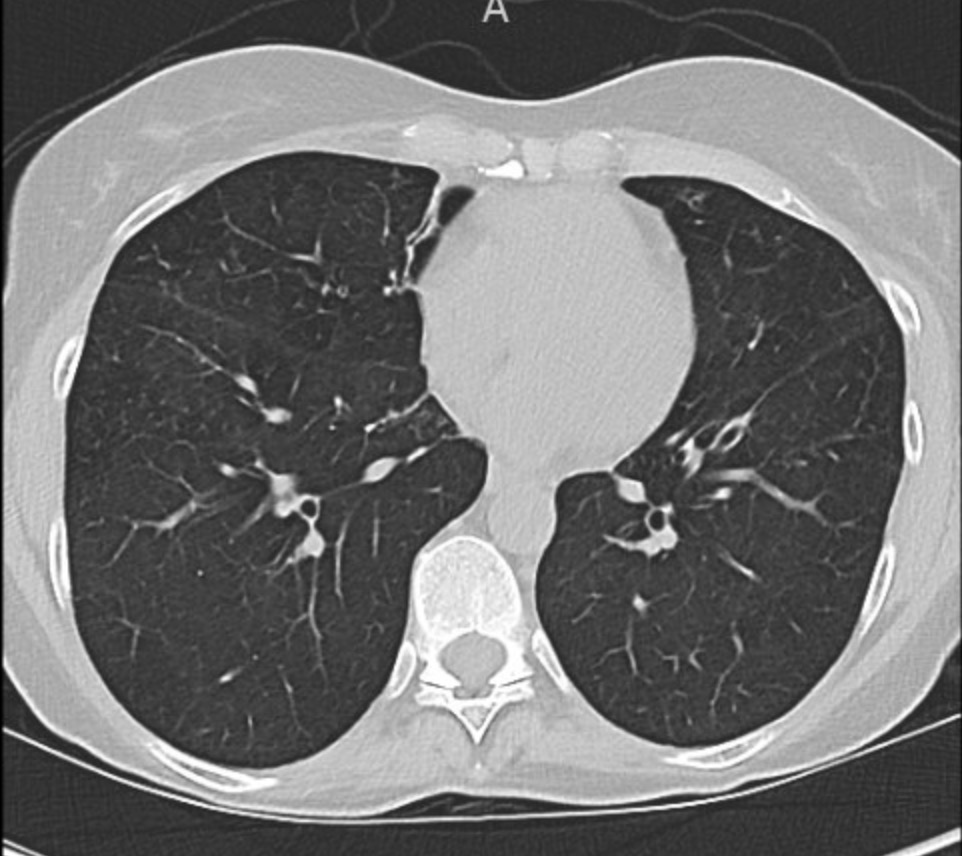
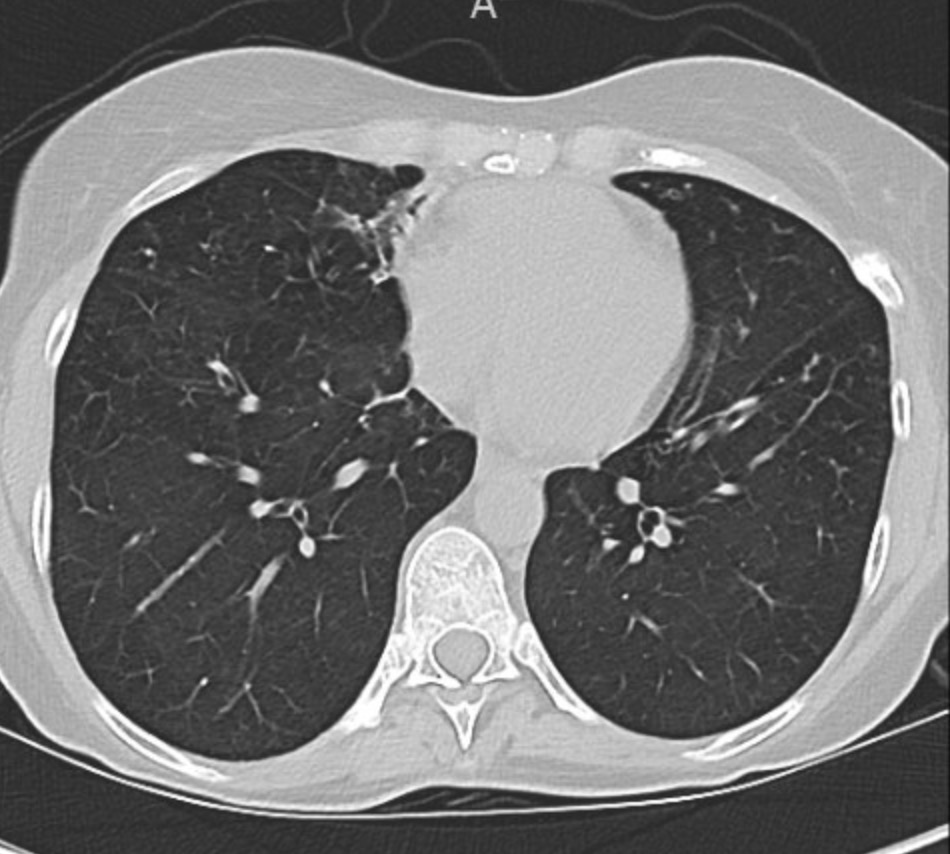
You can identify hyperinflation, interstitial changes and bronchial wall thickening on this image with concern for associated bronchiectasis
You should have Sarcoidosis, Cystic Fibrosis (CF), Hypersensitivity Pneumonitis (HP) and Lymphangioleiomyomatosis (LAM) on the differential
A CT chest is obtained given his abnormal chest x-ray and representative images are shown below
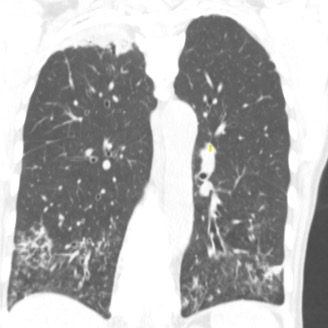
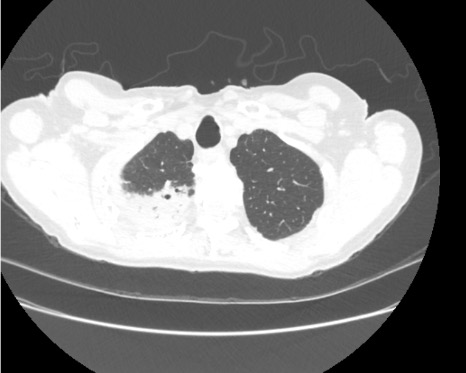
Given upper lobe bronchiectasis you are concerned for cystic fibrosis. A sweat chloride test is obtained and was elevated suggesting CF and further genetic testing was sent to confirm the diagnosis.
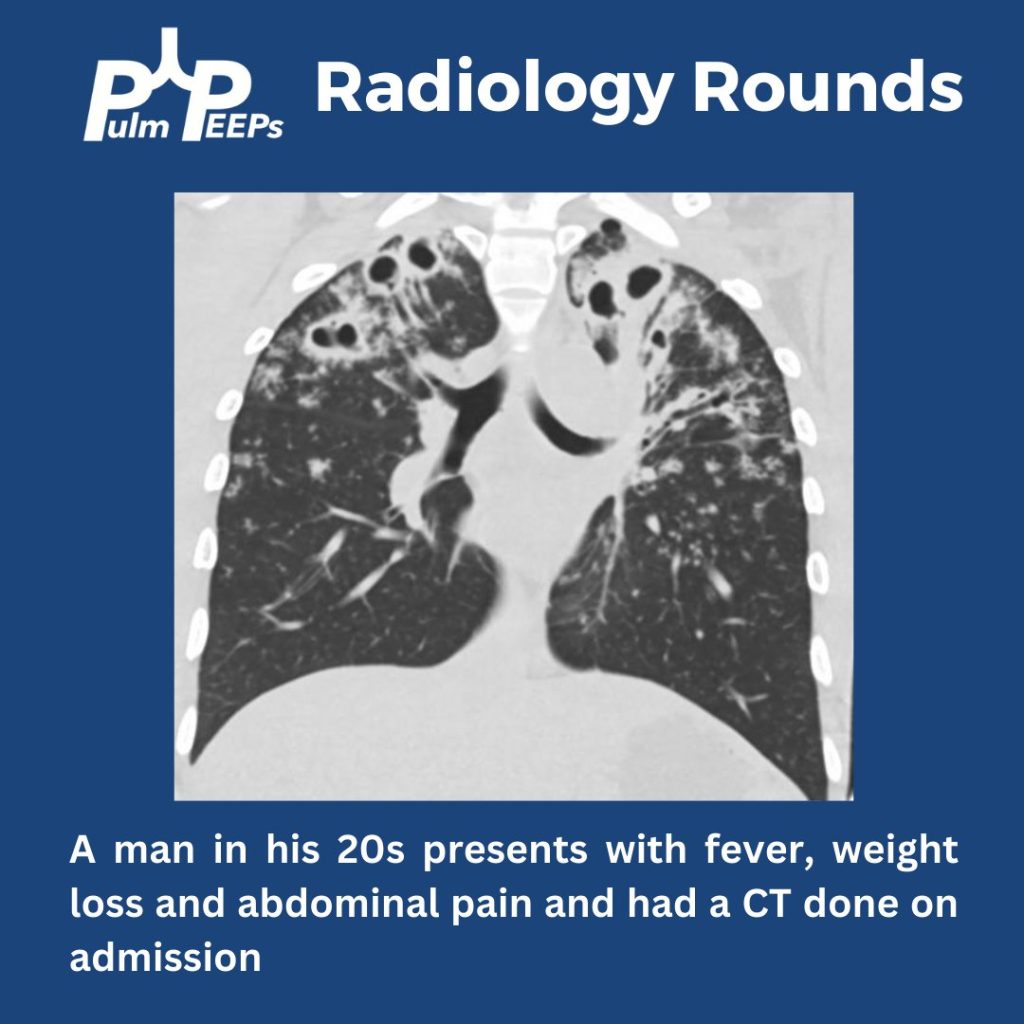
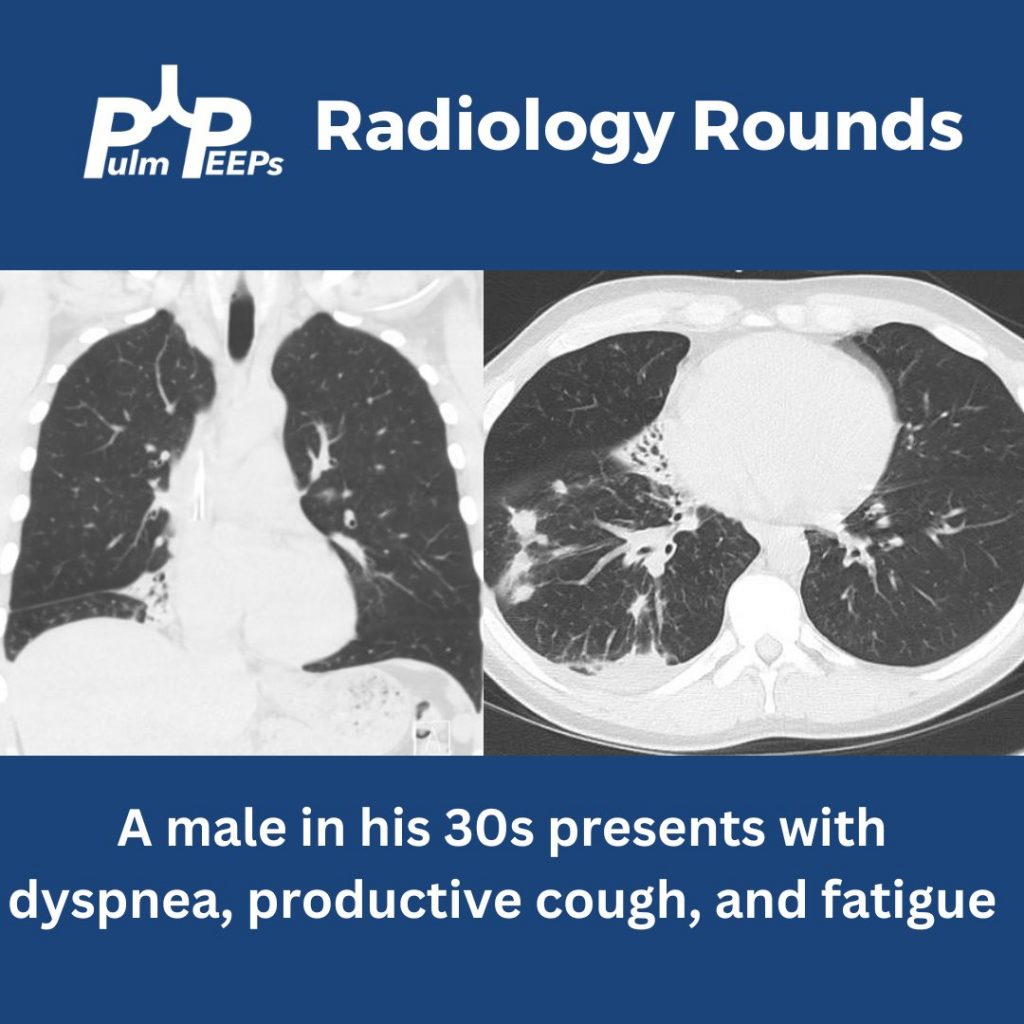
We are back with our first #RadiologyRounds of the new academic year. We have a young, immunocompetent man presenting with fever, weight loss, and abdominal pain.
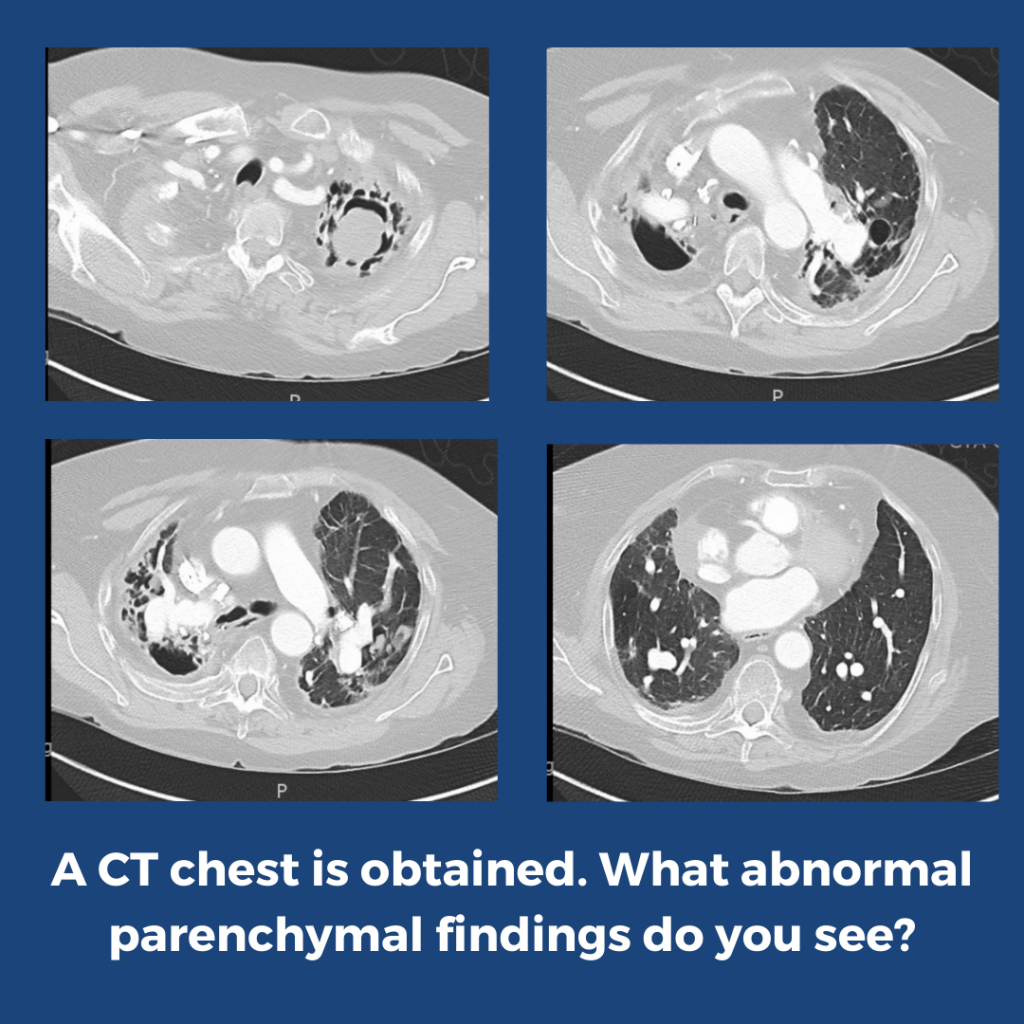
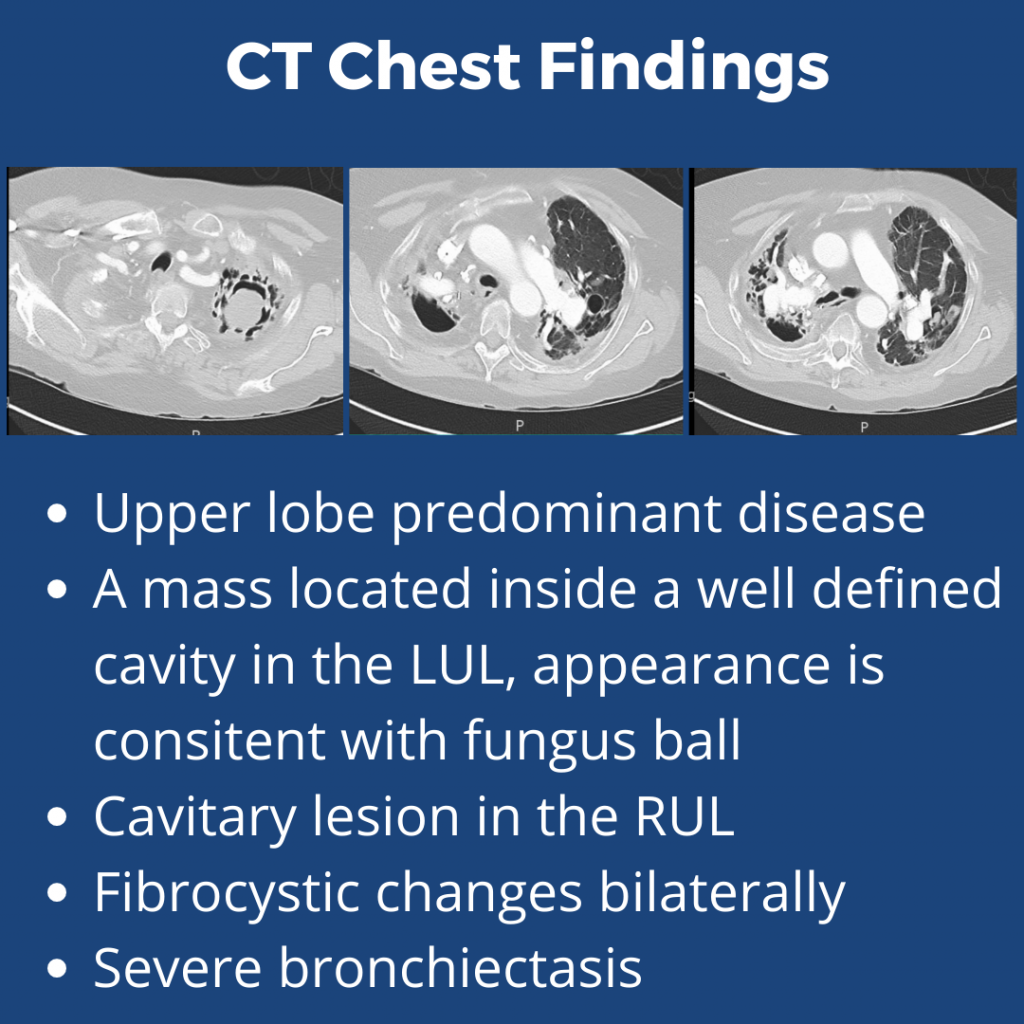
What abnormalities are seen on his chest imaging?
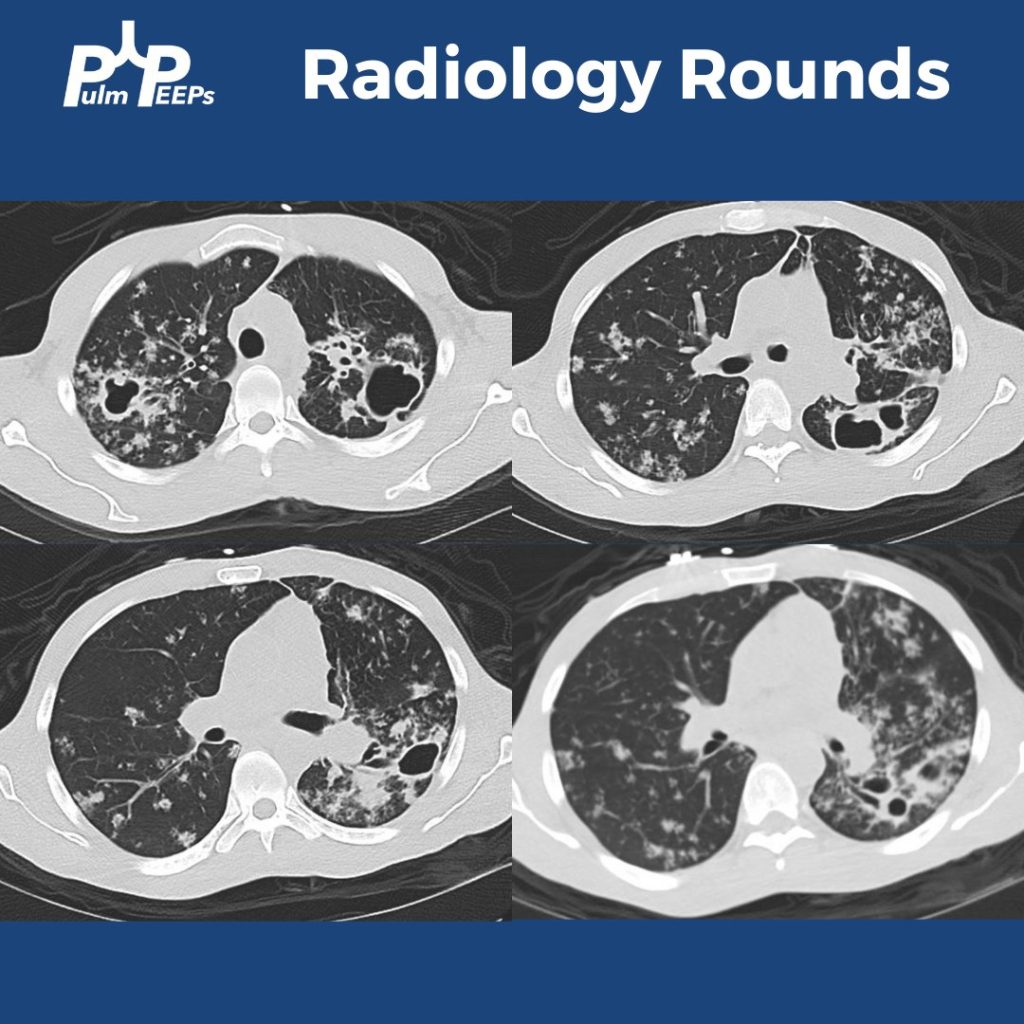
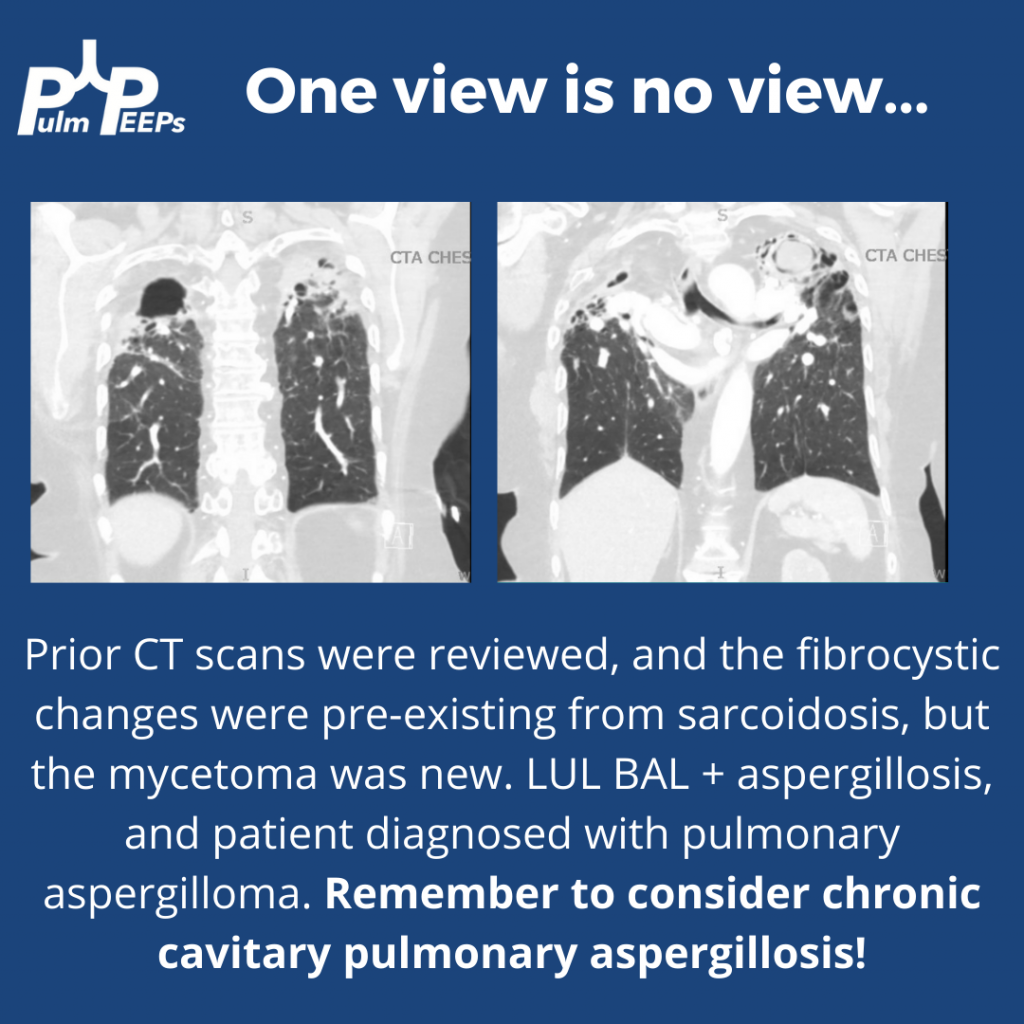
He was found to have bilateral apical cavitary disease, centrilobular nodules, and tree-in-bud opacities. He developed a productive cough with blood-tinged sputum as well as diarrhea.
Given his apical lung disease, what is on your differential?
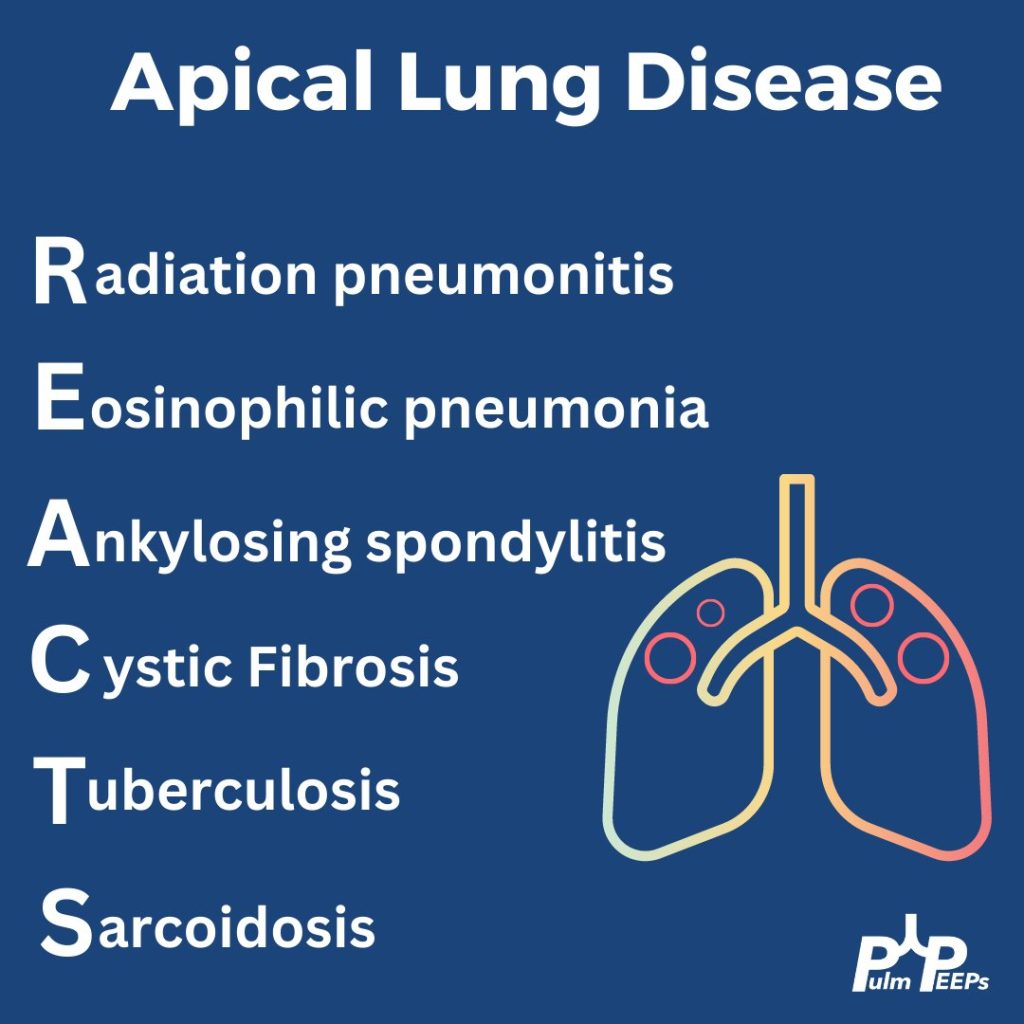
When thinking about apical lung disease, remember the mnemonic REACTS to help with your differential.
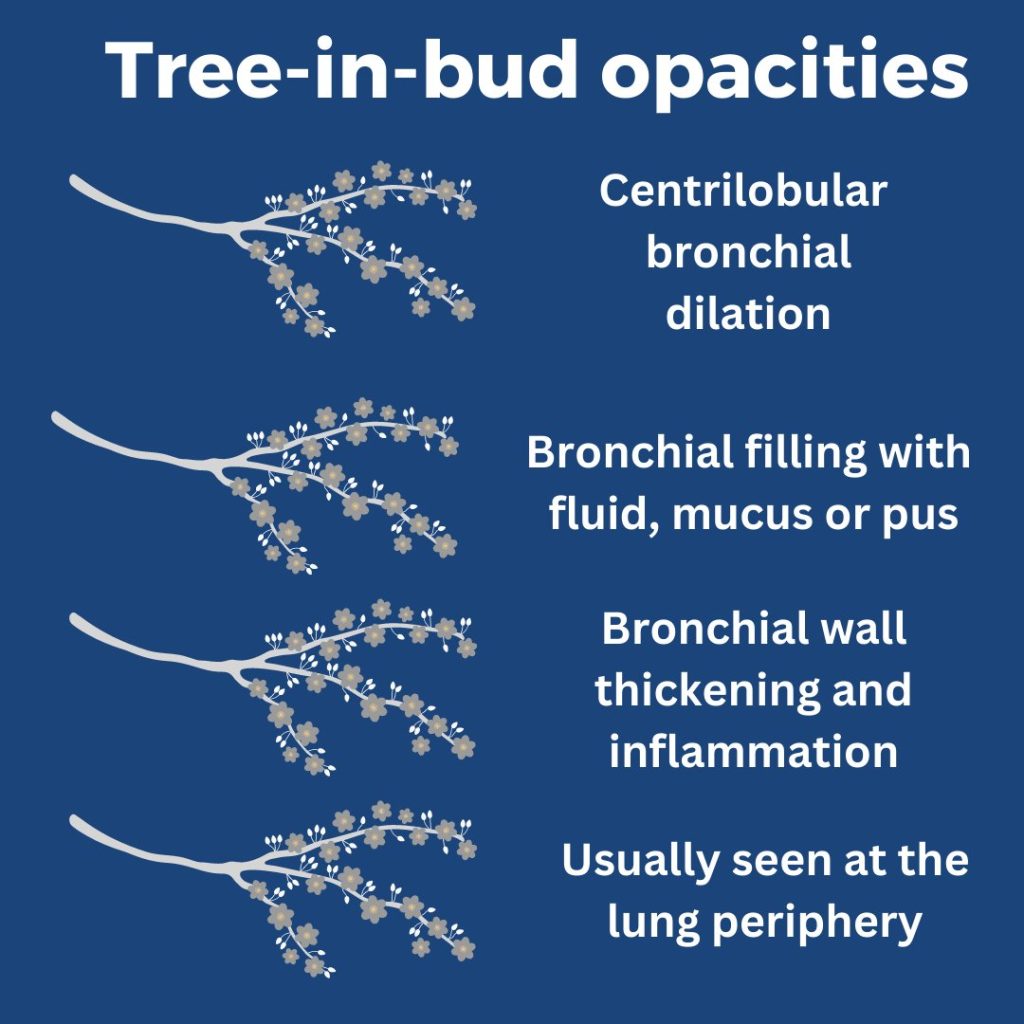
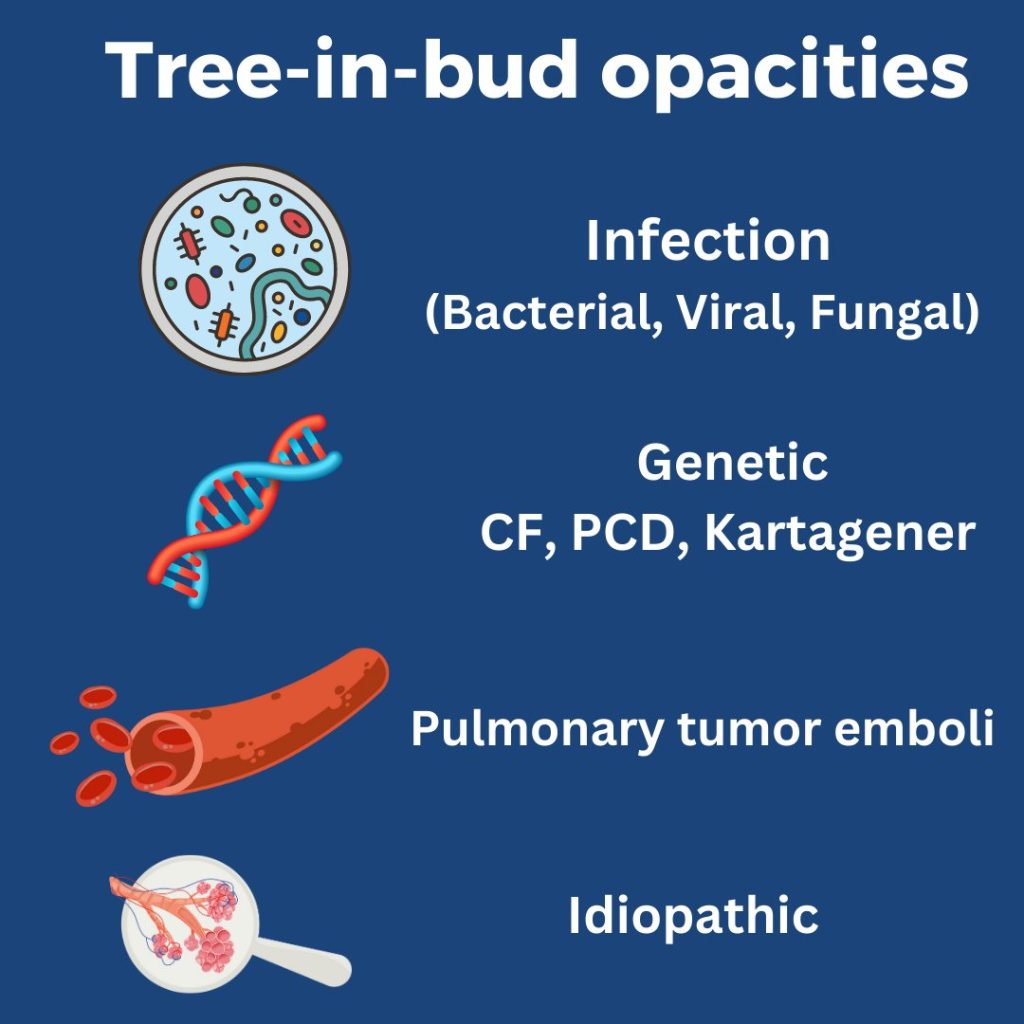
What are tree-in-bud opacities? They are findings seen on CT chest suggesting bronchial dilation, inflammation, and bronchial filling with fluid, mucus, or pus that can be caused by infections and non-infectious etiologies.
He had sputum and AFB cultures sent and his AFB smear was positive. He was ultimately diagnosed with disseminated TB and started on RIPE therapy.
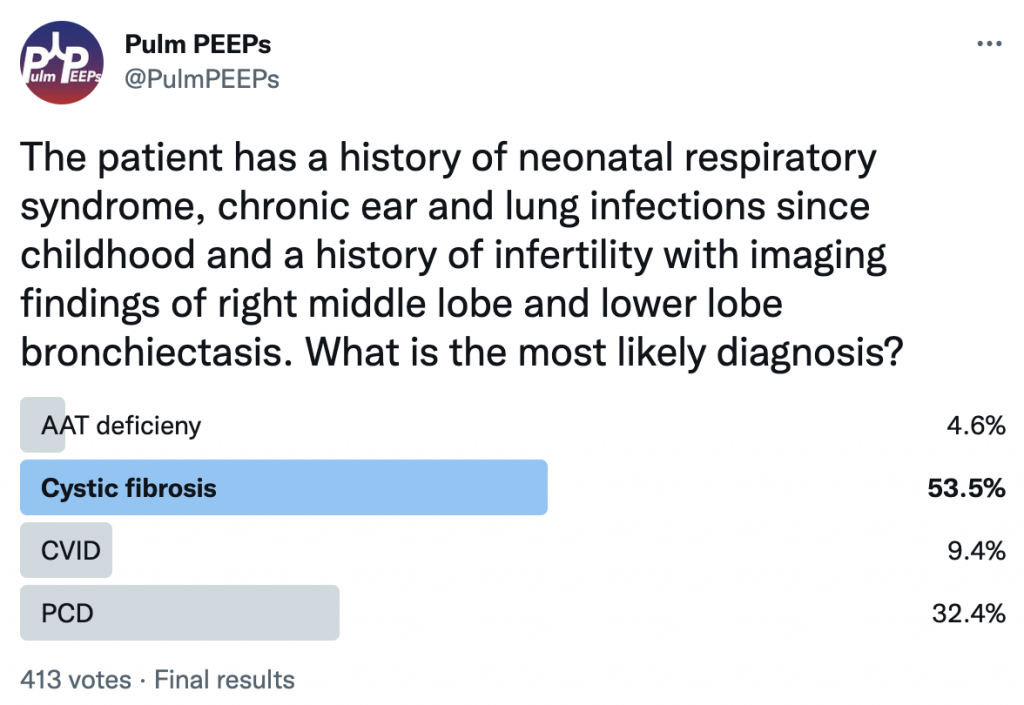
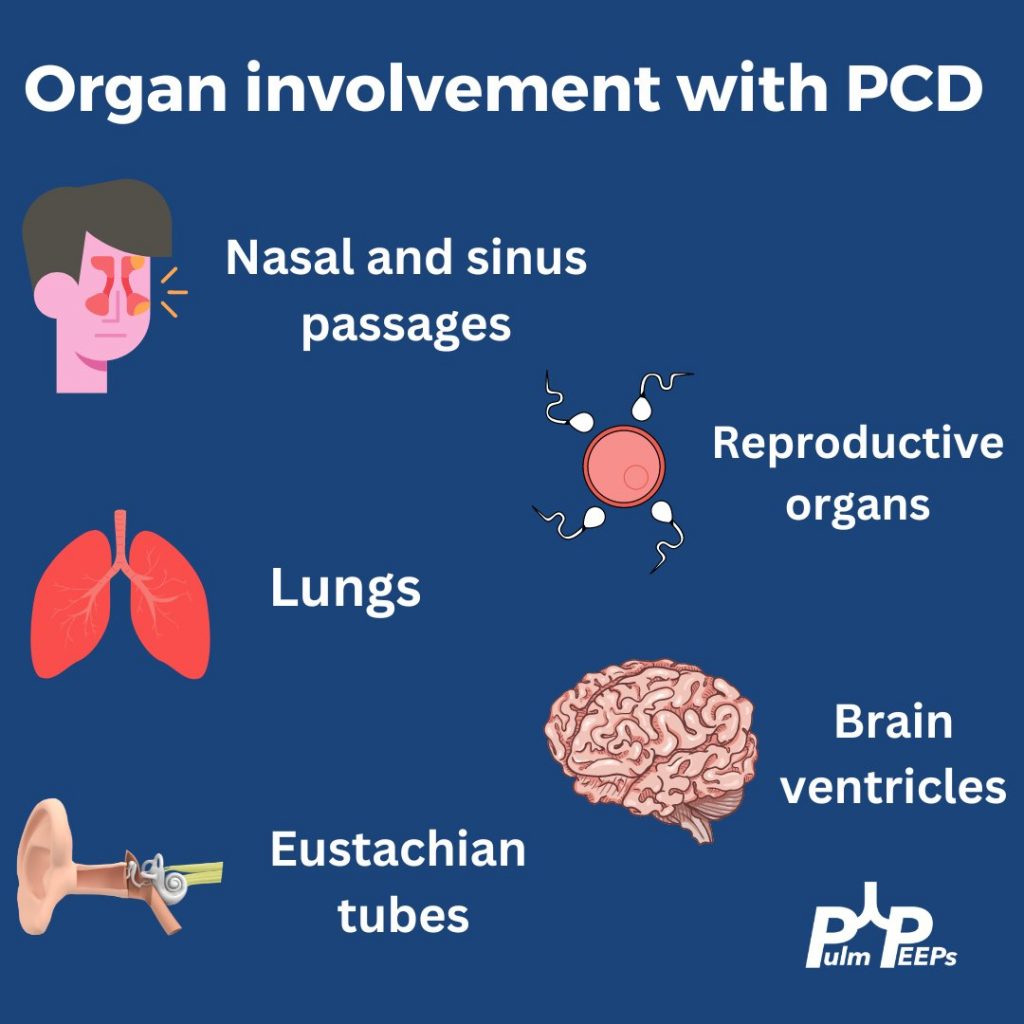
While chronic lung infections and infertility are overlapping symptoms for CF and Primary Ciliary Dyskinesia, the history of neonatal respiratory syndrome, ear infections and lower lobe bronchiectasis are most consistent with PCD
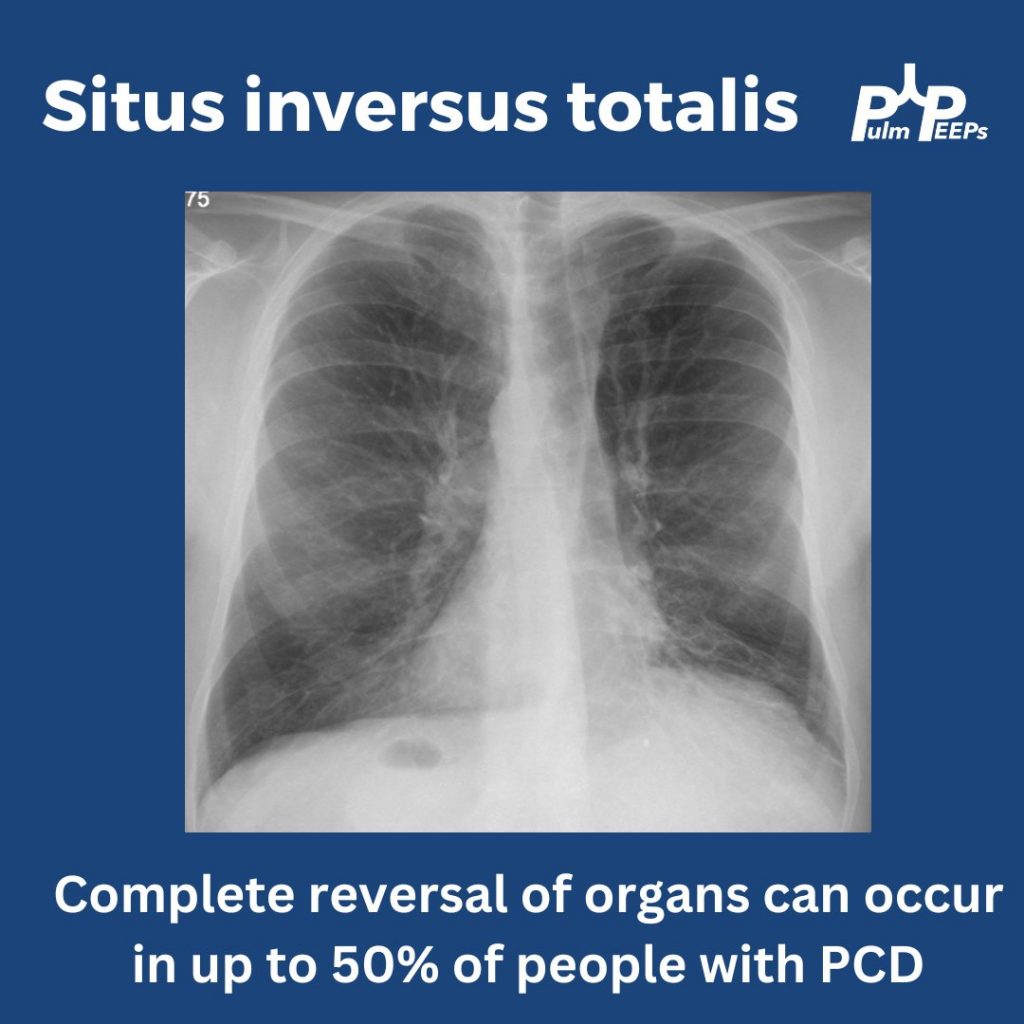
In up to 50% of people with PCD, you can get complete reversal of thoracic and abdominal organs. In this film you can see the heart in the right hemithorax, the gastric bubble on the right with the liver on the left resulting in elevation of the left hemidiaphragm
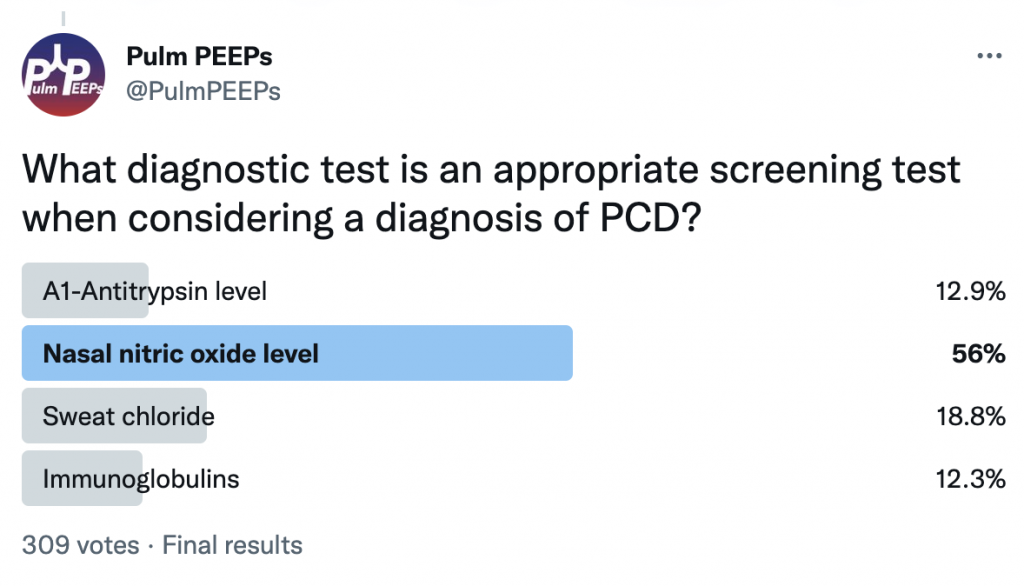
In patients with clinical symptoms and two decreased levels of nasal nitric oxide these findings suggest a PCD diagnosis but evaluation of the cilia structure and function as well as genetic testing are other diagnostic evaluations to confirm a diagnosis of PCD
Primary ciliary dyskinesia is a autosomal recessive disorder that results in motile ciliary dysfunction and clinical manifestations can vary depending on which organs are involved
This week’s #RadiologyRounds is authored by Leon Mirson, one of our amazing Associate Editors. Follow us on Twitter to answer live polls about the case and future Radiology Rounds cases!
The Pulm PEEPs are joined again by Natalie West to discuss a patient who presented with a chronic, productive cough. Listen in today as we work through our differential diagnosis, interpret basic pulmonary testing, and share our clinical reasoning along the way. We have some fantastic diagnostic and treatment teaching points, so once you’ve solved the case check out the takeaways and infographics below. Please let us know any additional insights you have on Twitter!
Patient Presentation
A 50-year-old woman, who is a never smoker, with a past medical history of recurrent pancreatitis presents to the pulmonary clinic with a chronic, productive cough. Her cough has been present for 3 years and has increased in frequency to now being present daily. In the last three months, the cough has also worsened and is productive of small amounts of yellow to green sputum. She has a history of chronic post-nasal drip and sinus infections, and uses intranasal steroids, but has not noted changes in these symptoms. There is no significant family history of pulmonary disease, and an exposure history review of symptoms is negative.
On physical exam, she was a thin woman who appeared her stated age and was breathing comfortably on room air. Her exam was notable for mild expiratory wheezing, primarily on auscultation of the right posterior lung field. She had no cyanosis, clubbing, evidence of volume overload, or abdominal tenderness.
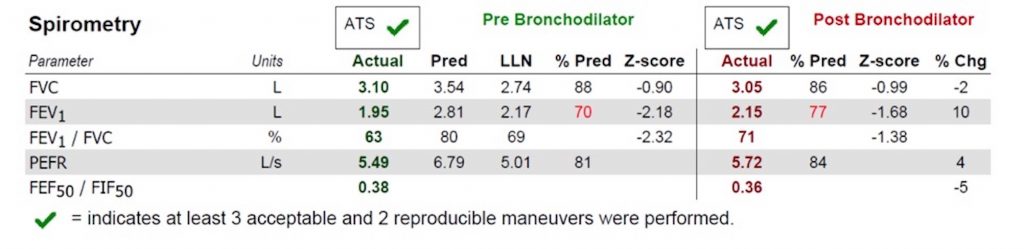
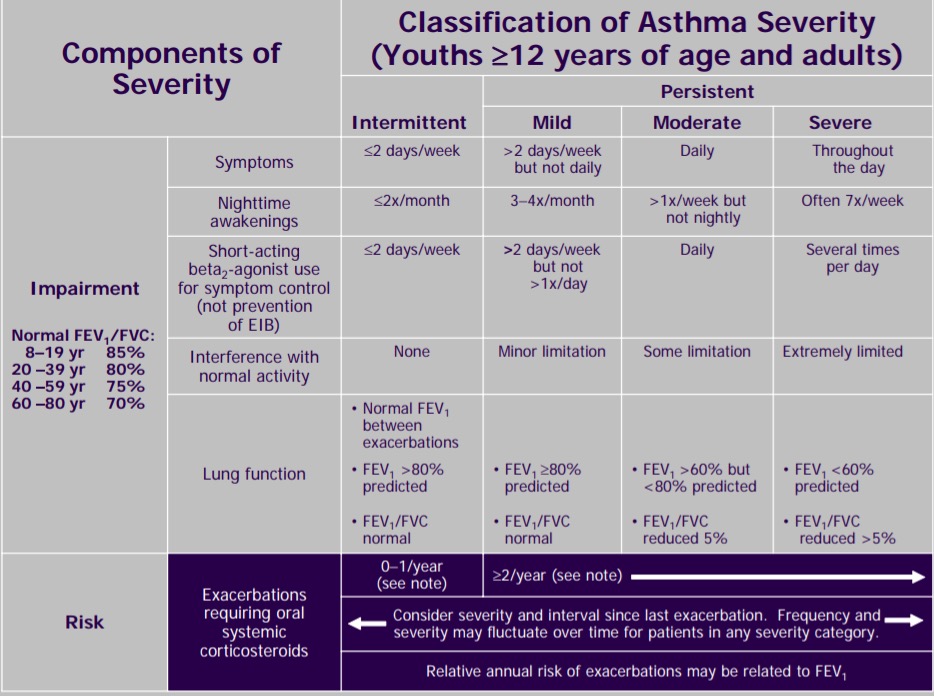
Basic Spirometry Values
Chest X-ray
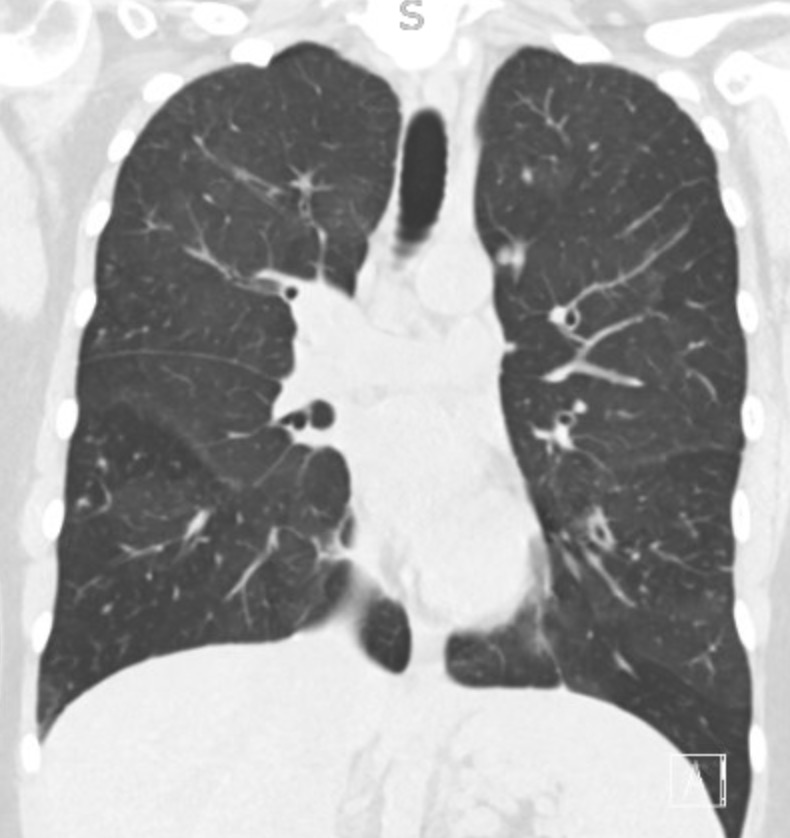
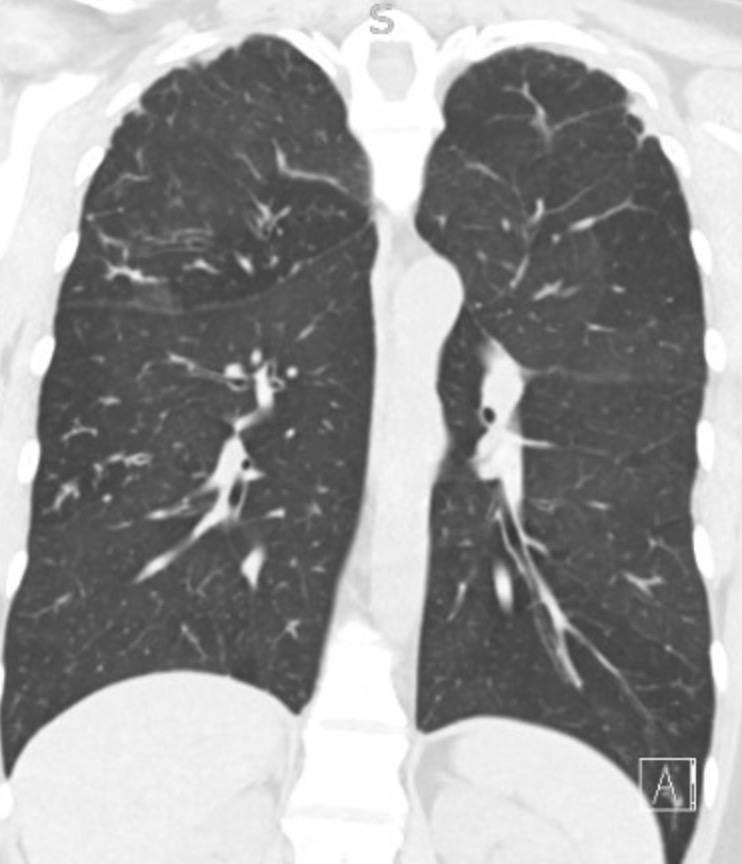
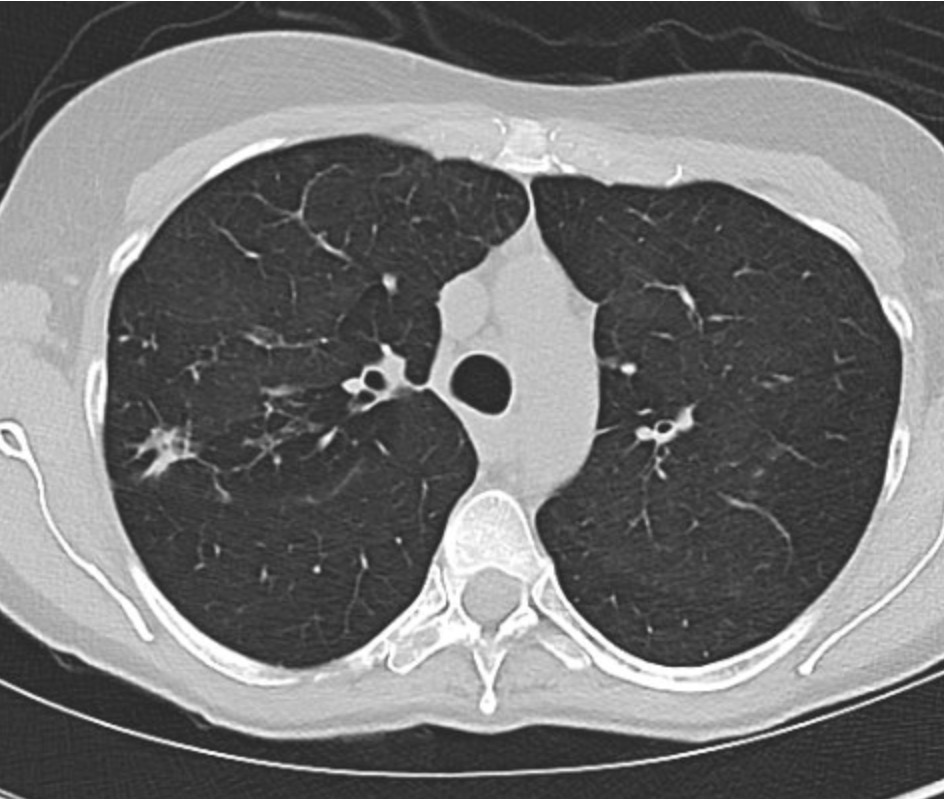
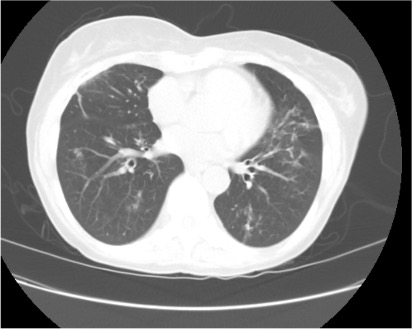
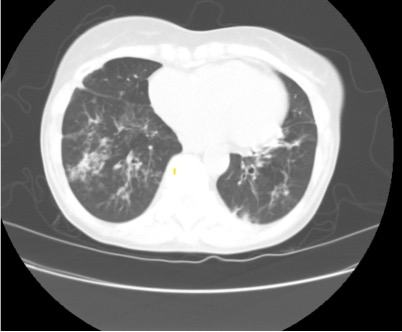
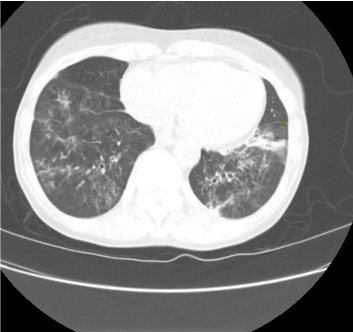
Representative Images from CT Scan
Key Learning Points
**Spoilers Ahead** If you want to think through the case on your own we advise listening to the episode first before looking at the infographics below.
Differential Diagnosis of Chronic Cough
Three most common causes: upper airway cough syndrome, GERD, cough variant asthma
Additional etiologies to consider: chronic bronchitis, post-infectious after a respiratory tract infection, bronchiectasis, ACE inhibitors, lung cancer, eosinophilic bronchitis, interstitial lung disease
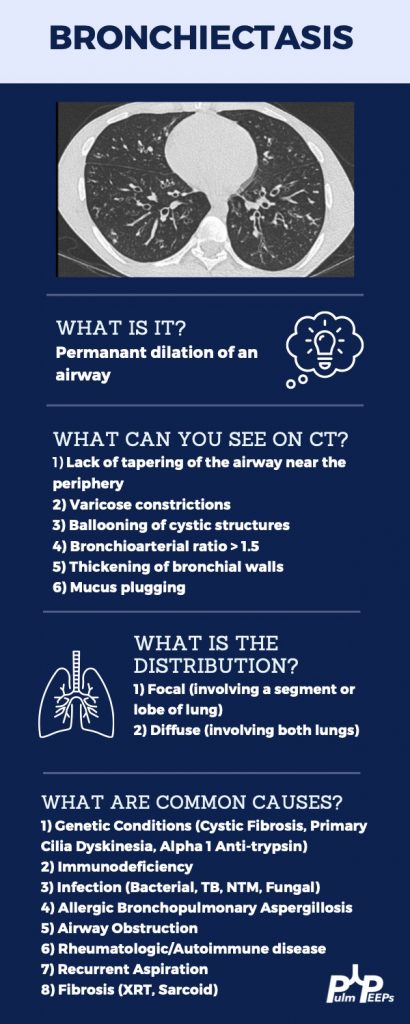
Imaging Pearl
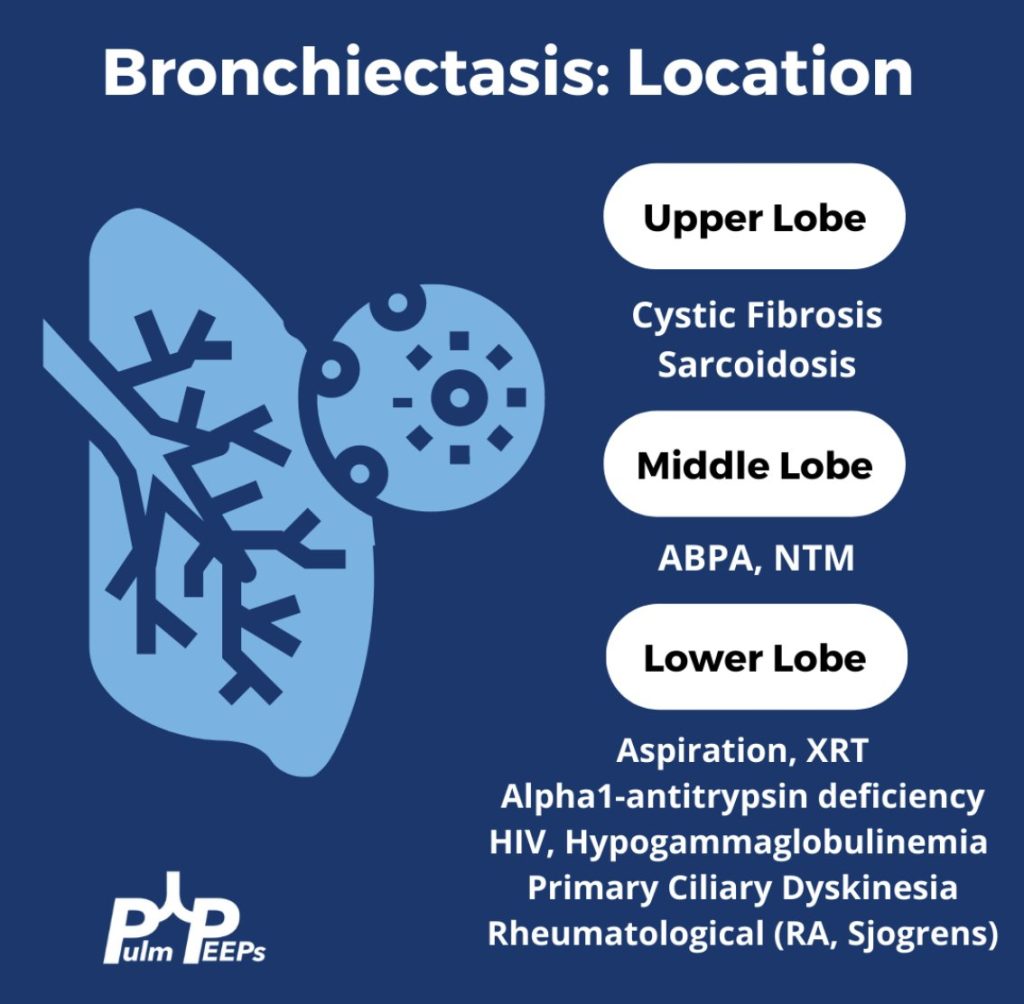
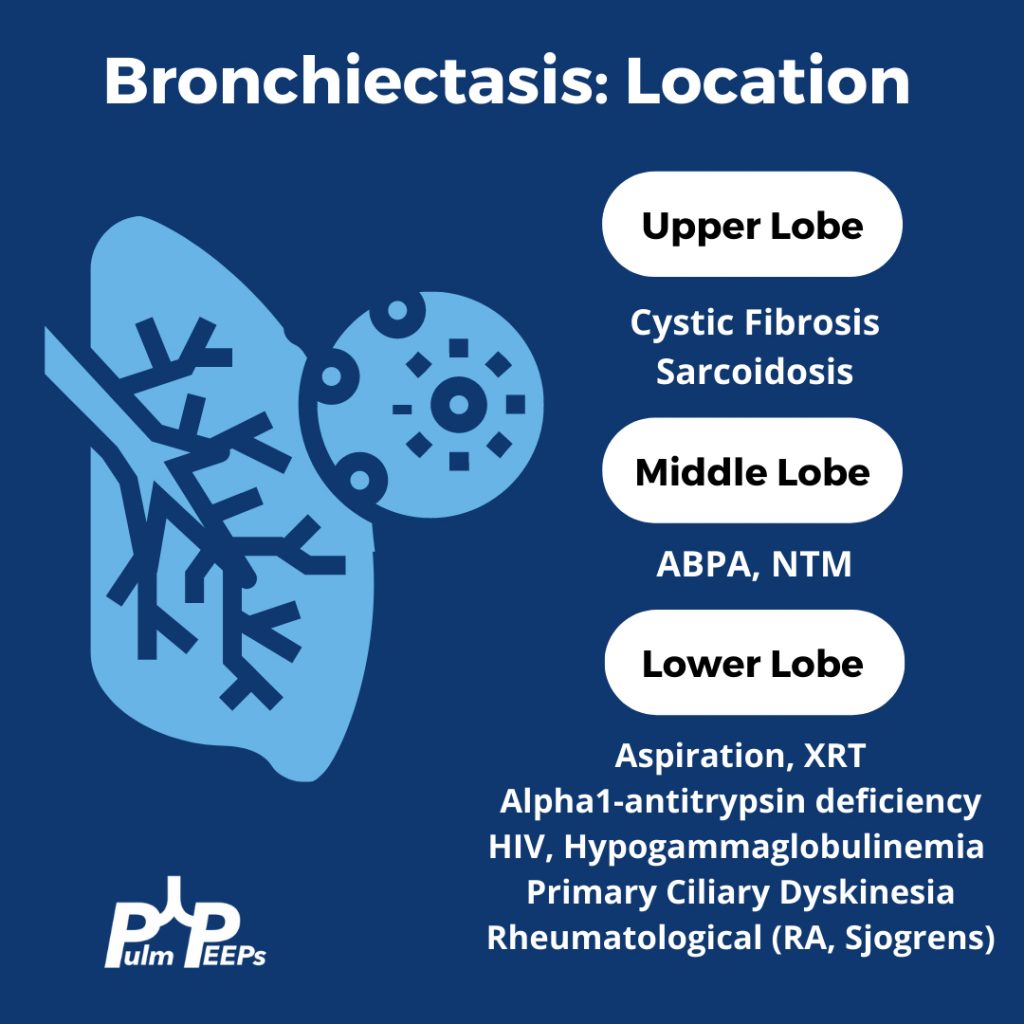
Evaluating Bronchiectasis
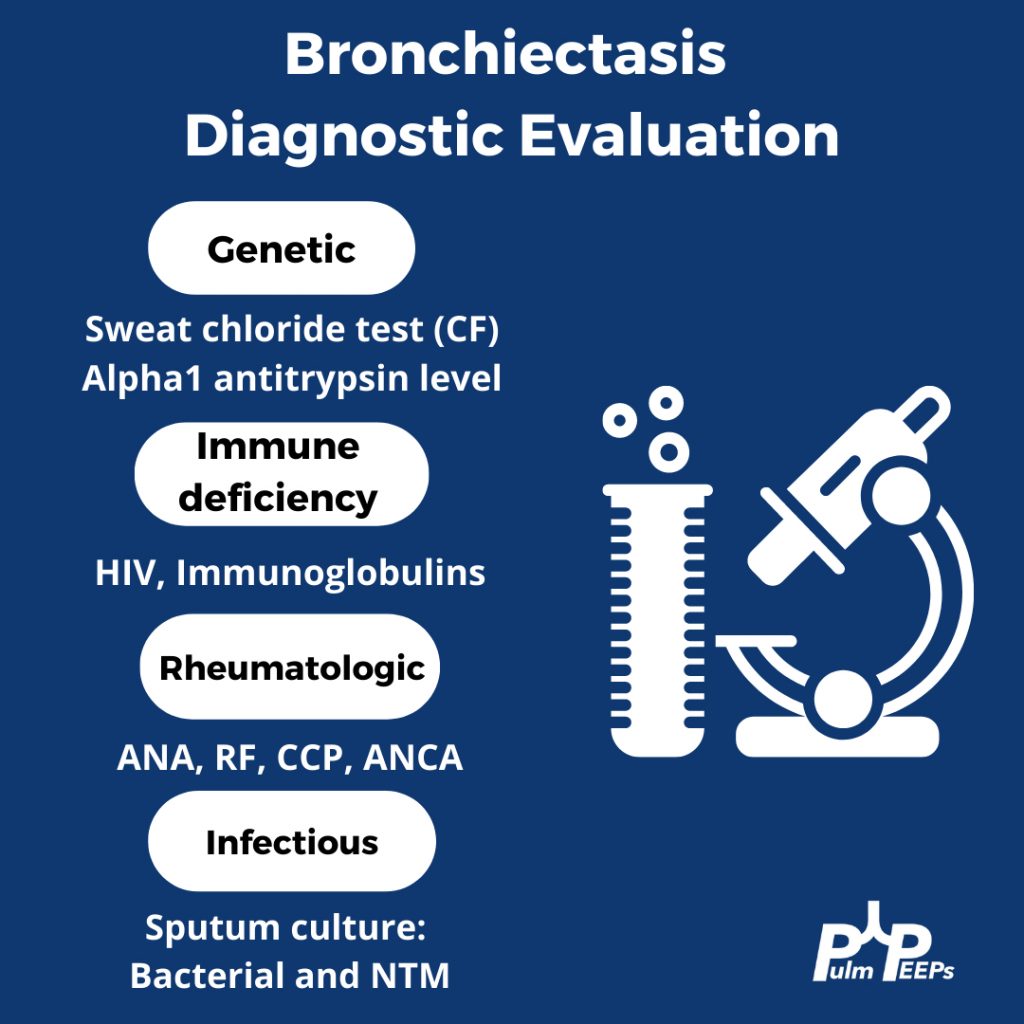
Making a New Diagnosis of Cystic Fibrosis in an Adult
Sweat testing
Sweat testing should be done in CF accredited center. Inform patients that there are no needles involved. Pilocarpine and electrical stimulation are applied to the arm or leg to stimulate the sweat gland, and then sweat is collected on filter paper, a gauze, or a plastic coil. From there, the amount of chloride in the sweat is calculated
Results
< 30 normal
31 – 60 indeterminate
> 60 is positive and Cystic Fibrosis is likely
What do you do with an Indeterminate test?
Patients with milder phenotypes of Cystic Fibrosis can have a normal or indeterminate sweat chloride level, and 10% of adults diagnosed with CF have a normal sweat chloride. If the sweat chloride test is indeterminate or normal, but suspicion is high for CF, then genetic testing for the whole array of mutations should be performed
The Pulm PEEPs are excited to bring our first mystery case! Kristina Montemayor and Dave Furfaro hear a fascinating case presentation from Pulm PEEPs senior editor Ansa Razzaq. Join us as we work through this case together to come to a diagnosis, and share our thought process along the way. Come back to these show notes afterward, or once you’ve solved the case yourself, for some key teaching pearls and representative images.
Patient Presentation
A 66-year-old woman with no smoking history and past medical history of previously well-controlled asthma is referred to pulmonary clinic after multiple recent episodes of dyspnea, wheezing, and coughing. The episodes have features consistent with asthma exacerbations; however, they are also associated with migratory infiltrates. She has been treated with multiple courses of antibiotics and steroids, and despite escalating therapy, the episodes are occurring more frequently and she was worsening overall exercise tolerance. Listen in to hear more and try to solve the case!
Representative Imaging
Key Learning Points
**Spoilers Ahead** If you want to think through the case on your own we advise listening to the episode first before looking at the infographics below.
The Pulm PEEPs (Kristina Montemayor and Dave Furfaro) host a panel of Cystic Fibrosis (CF) providers to discuss the current state of the disease, recent advances in cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapies, and the evolving faces and voices of Cystic Fibrosis.
Cystic Fibrosis is an autosomal recessive disorder caused by mutations in the CFTR gene that affects over 30,000 individuals in the United States and 70,000 people worldwide. Absence or dysfunction of the CFTR protein leads to abnormal secretion of mucus, sweat, and digestive fluids, which impacts the lungs, digestive tract, and reproductive system.
From the first formal publication on Cystic Fibrosis in 1938 by Dorothy Hansine Andersen, to the discovery of the delta F508 mutation and CFTR gene in 1988 -1989 by Lap-Chee Tsui, Francis Collins, and John R. Riordan, to the approval of the first CFTR modulator therapy, Ivacaftor, in 2012, our knowledge about Cystic FIbrosis has been advancing in leaps and bounds. As therapies have improved, they have dramatically impacted the lives of patients with Cystic Fibrosis. Join us today as we explore what this evolution in care has looked like from the perspective of Cystic Fibrosis providers, and hear about the new questions and challenges on the horizon.
Meet our guests
Emily DiMango is a Professor of Medicine at Columbia University Medical Center and the Director of the John Edsall-John Wood Asthma Center and the Gunnar Esiason Adult Cystic Fibrosis Program
Terri Laguna is an Associate Professor of Pediatrics at Northwestern Medicine / Feinberg School of Medicine and the Chief of Pulmonary and Sleep Medicine in the Department of Pediatrics
Patrick Sosnay is a Senior Medical Director at Vertex Pharmaceuticals and specializes in Cystic Fibrosis
Natalie West is an Assistant Professor of Medicine at Johns Hopkins Hospital and specializes in Cystic Fibrosis.